Download
1 / 39
430 likes | 851 Views
Sickle Cell Anemia. Gregg Selke, Ph.D. 11/28/06. What is Sickle Cell Anemia (SCA)?. First described in Chicago in 1910 by James Herrick as an inherited condition that results in a decrease in the ability of red blood cells to carry oxygen throughout the body.

E N D
Sickle Cell Anemia Gregg Selke, Ph.D. 11/28/06
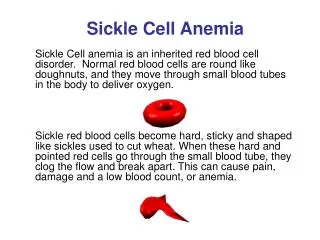
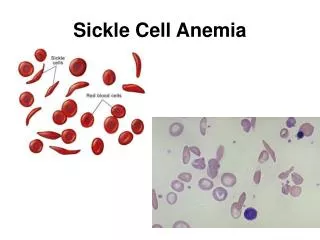
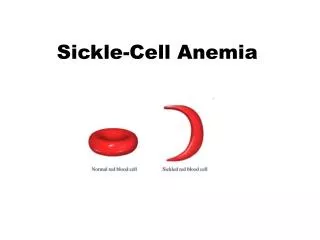
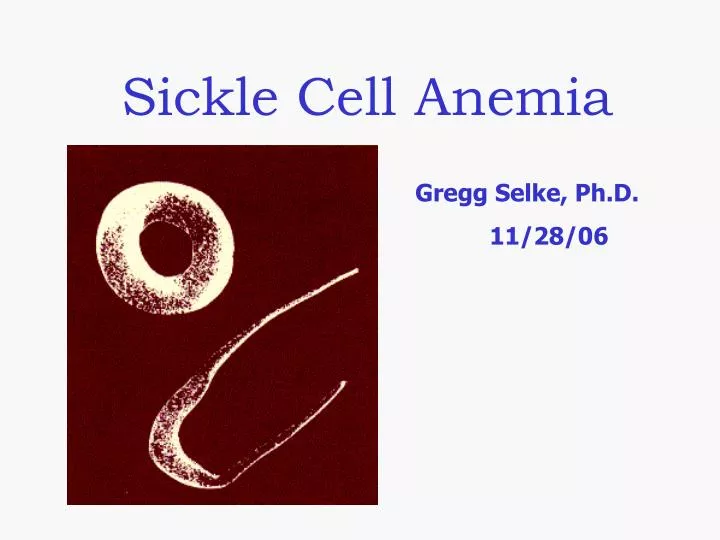
What is Sickle Cell Anemia (SCA)? • First described in Chicago in 1910 by James Herrick as an inherited condition that results in a decrease in the ability of red blood cells to carry oxygen throughout the body • Sickle red blood cells become hard and irregularly shaped (resembling a sickle) • Become clogged in the small blood vessels and therefore do not deliver oxygen to the tissues. • Lack of tissue oxygenation can cause excruciating pain, damage to body organs and even death.
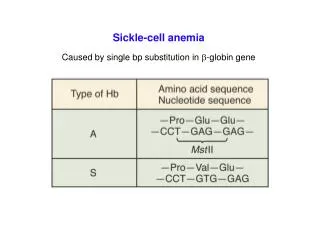
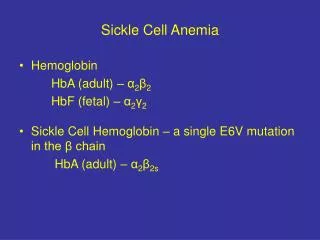
Mechanism • Red blood cells (RBC) • Contain a special protein called haemoglobin (Hb) • Hb is the component that carries oxygen from the lungs to all parts of the body • Most people have only hemoglobin type – Hb A within RBC (normal genotype: Hb AA) • Sickle Cell: HbS • S similar to A, but one structural change • Other types: HbC, HbD, and HbE
Mechanism -HbS • When sickle haemoglobin (HbS) gives up its oxygen to the tissues, HbS sticks together • Forms long rods form inside RBC • RBC become rigid, inflexible, and sickle-shaped • Unable to squeeze through small blood vessels, instead blocks small blood vessels • Less oxygen to tissues of body • RBCs containing HbS have a shorter lifespan • Normally 120 days • Chronic state of anaemia
Genetics • 2 copies of the gene for Hb (each parent) • HbS –Recessive • S=Sickle • A=Normal
Sickle Cell Trait • Sickle haemoglobin (S) + Normal haemoglobin (A) in RBC • Adequate amount of normal Hb (A) in red blood cells • RBC remain flexible • Carrier • Do Not have the symptoms of the sickle cell disorders, with 2 exceptions • Pain when Less Oxygen than usual (scuba diving, activities at high altitude (12,000ft), under general anaesthesia) • Minute kidney problems
Three common types of Sickle Cell Disorders • Sickle Cell Anemia • Sickle haemoglobin (HbS) + Sickle haemoglobin (HbS) • Most Severe – No HbA
Other Sickling Disorders Other types of Hb combine with sickle Hb • Hemoglobin S-C disease • Sickle haemoglobin (HbS) + (HbC) • Hemoglobin S-Beta thalassemia • Beta thalassaemia gene reduces the amount of HbA that can be made • Sickle haemoglobin (HbS) + reduced HbA • Milder form of Sickle Cell Disorder than sickle cell anemia
Some Genetic History • The error in the hemoglobin gene results from a genetic mutation that occurred many thousands of years ago in people in parts of Africa, the Mediterranean basin, the Middle East, and India. • A deadly form of malaria was very common at that time • Malaria epidemics caused the death of many • In areas where malaria was a problem, children who inherited one sickle hemoglobin gene and who, therefore, carried the sickle cell trait - had a survival advantage. • Unlike the children who had normal hemoglobin genes, they survived the malaria epidemics they grew up, had their own children, and passed on the gene- for sickle hemoglobin.
Sickle Cell Gene Severe Malaria
History • As populations migrated, the sickle cell-mutation spread to other Mediterranean areas, further into the Middle East and eventually into the Western Hemisphere. • In the United States and other countries where malaria is not a problem, the sickle hemoglobin gene no longer provides a survival advantage. • Instead, it may be a serious threat to the carrier's children, who may inherit two abnormal sickle hemoglobin genes and have sickle cell anemia.
Who is at risk? • Most common in Africans and African Americans. • East Asia, Southern Italy, Saudi Arabia, India, Egypt, South and Central American, Cuba, the Caribbean, Greece, and Iran, and Eastern Jews have also been found to have a form of this illness.
Prevalence • More than 2.5 million Americans have the trait • 70,000 or more Americans have sickle cell disease • About 1,000 babies are born with the disease each year in America • In Nigeria, 1/3 population of U.S., 45,000-90,000 babies with sickle cell disease are born each year
Among African - Americans • 1 in 12 have Sickle Cell Trait (Hb SA) • 1 in 600 have Sickle Cell Anemia (Hb SS) • 1 in 1500 have Sickle C Disease (Hb SC) • 1 in 350 have Sickle Cell Disease (Hb SS, SC, S-Beta-Thal) • Among Latinos • 1 in 172 have Sickle Cell Trait (Hb AS) • 1 in 1,000 have Sickle Cell Disease (Hb SS, SC, S-Beta-Thal)
Screening • Haemoglobin Electrophoresis • Simple Blood test • Routine screening in high risk groups • During pregnancy • Before anaesthesia • Prenatal Testing • Amniocentesis • 16 and 18 weeks of the pregnancy • small risk of causing a miscarriage (1 in 100) • Chorionic villus sampling (CVS) • 9th or 10th week of pregnancy • very small amount of material from the developing placenta • slightly higher chance of miscarriage
Early Symptoms and Complications • Typically appear during infant's first year • 1st symptom: dactylitis and fever (6 mo-2 yrs) • Pain in the chest, abdomen, limbs and joints • Enlargement of the heart, liver and spleen nosebleeds • Frequent upper respiratory infections • Chronic anemia as children grow older • Over time Sickle Cell sufferers can experience damage to organs such as liver, kidney, lungs, heart and spleen • Can result in death
Medical Complications • kidney damage and • loss of body water in urine • painful erections in men (priapism) • blood blockage in the spleen or liver (sequestration) • eye damage • low red blood cell counts (anemia) • delayed growth • pain episodes • strokes • increased infections • leg ulcers • bone damage • yellow eyes or jaundice • early gallstones • lung blockage
Serious Complications • Infectious complications • Prominent early in life • Leading cause of morbidity and mortality • Great improvement in the prognosis related to newborn screening for sickle cell disease, vaccination for childhood illnesses, the use of prophylactic antibiotics, and aggressive diagnosis and treatment of febrile events • Acute splenic sequestration • Episodes of rapid increase in splenic size and decrease in hemoglobin • Potential source of morbidity and mortality early in life for children with sickle cell anemia and at any age for those with Hb SC disease and sickle thalassemia
Serious Complications • Strokes • Up to 15% of children may have overt or silent strokes during childhood • Chronic transfusion therapy reduces the recurrence rate of overt stroke which may approach 75% without intervention • Bone disease • Early risk is primarily from osteomyelitis • Infectious usually painful inflammatory disease of bone often of bacterial origin and may result in bone tissuedeath • Avascular necrosis of the femur and humerus • Death of bone tissue due to disrupted blood supply • Marked by severe pain in the affected region and by weakened bone that may flatten and collapse
Serious Complications • Leg ulcers • Seen in patients older than 10 years of age • Resistant to therapy and cause significant morbidity • Ophthalmic complications • Proliferative retinopathy, vitreous hemorrhage, & retinal detachment • Priapism • Distressing complication that occurs at all ages • Difficult to treat • Causes a high incidence of impotence • Chronic Anemia • Associated with fatigue, irritability, jaundice, pain, delayed puberty, leg sores, eye problems, gum disease
Serious Complications: PAIN Recurrent Pain Episodes or Sickling Crises • Occur at any age but appear to be particularly frequent during late adolescence and early adult life • Unpredictable • Red Blood Cells get stuck in the small veins and prevent normal blood flow • Characterized by severe pain in the back, chest, abdomen, extremities, and head • Highly disruptive to life • Most common reasons for individuals to seek health care
Danger Signs of a Crisis • Any sudden weakness or • loss of feeling • Pain that will not go away • with home treatment • Priapism (painful erection • that will not go down) • Sudden vision change • Fever • Chest pain • Shortness of Breath • Increasing tiredness • Abdominal swelling • Unusual headache SEEK URGENT HOSPITAL TREATMENT IF IN CRISIS
Crises • During a crisis • severe pain in the fingers, toes, arms, joints,legs, back, abdomen, and bones. • Decrease in oxygen to the chest and lungs • May lead to acute chest syndrome • Damage to the lungs • Severe pain and fever • Lungs' airways narrow, further reducing O2 • Leads to an increased risk of potentially • fatal infections
Triggers of Pain • Infections • Thirst and dehydration caused by not drinking enough even if thirst is not felt • Over-exertion • Over-excitement • Cold weather and cold drinks and swimming • Bangs, bumps, bruises and strains • Stress triggers pain in adults, but does not seem to do so inchildren.
Predicting Pain • Children and families can often tell when a severe sickle pain is coming on by • Thirst • Eyes turning yellow (jaundice), • Sufferer being more irritable or tired than usual.
Alleviating Pain • Warmth: increases blood flow • Massaging and rubbing • Heat from hot water bottles and deep heat creams • Bandaging to support the painful region • Resting the body • Cognitive Behavioral Therapy • Getting the sufferer to relax • deep breathing exercises • distracting the attention • by other psychological methods. • Pain-killing medicines (analgesics): paracetamol, codeine non-steroidal anti-inflammatory, morphine if necessary
Daily Preventative Measures • Taking the folic acid (folate) daily to help make new red cells • Daily penicillin until age six to prevent serious infection • Drinking plenty of water daily (8-10 glasses for adults) • Avoiding too hot or too cold temperatures • Avoiding over exertion and stress • Getting plenty of rest • Getting regular check-ups from knowledgeable health care providers
Treating Complications • Pain-killing drugs and oral and intravenous fluids • To reduce pain and prevent complications. • Transfusions • Correct anemia • Treat spleen enlargement in children before the condition becomes life-threatening • Regular transfusion therapy also can help prevent recurring strokes in children at high risk of crippling nervous system complications.
Psychosocial Issues • Require regular medical attention • Especially before and after operations, dental extraction and during pregnancy. • Adherence to medical regimen • Vitamins, antibiotics, fluid intake, activity level • Schools must be involved • Family planning • Suitable types of employment • Air travel • Increased fluids, pain killers or oxygen may be recommended
Psychosocial Issues • Child should be encouraged to participate in sports, but not pushed passed their limitations • If they are in pain or feel tired they should be allowed to rest and keep warm. • They should have access to drinks. • Strenuous exercise, dehydration and cold can induce a crisis. • Strenuous outdoor activities should be avoided in cold or wet weather • Should only swim if the water is warm and care is taken to keep warm when leaving the water • If develops a crisis despite these precautions he or she should avoid swimming all together
Psychosocial Issues • Child Specific Issues: Coping with Pain • Pain happens more often • On an average of one third of all days • Lasts longer • Generally all day, even if not continuously all day • Associated with great tiredness about half the time • Causes them to spend significant time in bed • On average the time spent wholly or partly in bed adds up to about a week of every school term.
Psychosocial Issues • Variability and Unpredictability • Some are mildly affected and largely free from pain, while others have frequent and severe pain • Most children go through good and bad patches • Doctors cannot predict who will be severely affected. • No easily overt detectable signs of sickle pain • So children known to have sickle cell disorder who say they are in pain must be trusted • If they can rely on the adults around them to take them seriously, they are less likely to take advantage of their condition to seek attention or avoid distasteful tasks.
Psychosocial Issues • To reduce risk of crisis, children are encouraged to drink much more than normal and more frequently • May require about 1/4 litre of liquid every 60 - 90 minutes. • Child will need to go to the toilet more frequently • May increase risk of Enuresis • Boys at risk for priapism • May be too embarrassed to mention to parents • Severe sickling can lead to impotence
Developing Treatments • Hydroxyurea • The first effective drug treatment for adults with severe sickle cell anemia reported in early 1995 • Daily doses of the anticancer drug, hydroxyurea, reduced the frequency of painful crises, acute chest syndrome, needed fewer blood transfusions • Increases production of fetal hemoglobin in the blood • Fetal hemoglobin seems to prevent sickling of red cells • cells containing fetal hemoglobin tend to survive longer in the bloodstream
Developing Treatments • Bone marrow transplantation • Shown to provide a cure for severely affected children with sickle cell disease • Only about 18 percent of children with sickle cell anemia are likely to have a matched sibling.
The Ultimate Cure? • Gene Therapy • Correcting the “defective gene” and inserting it into the bone marrow • Turning off the defective gene and simultaneously reactivating another gene that turns on production of fetal hemoglobin. • No real cure for Sickle Cell Anemia at this time. • “In the past 30 years, the life expectancy of people with sickle cell anemia has increased. Many patients with sickle cell anemia now live into their mid-forties and beyond.”
Websites http://www.sicklecellsociety.org/ : Another Great Site information, Counselling and Caring for those with Sickle Cell Disorders and their families: UK based http://www.sicklecelldisease.org/: Sickle Cell Disease Association of America The Human Genome Project Sickle Cell Education Site at http://www.massinteraction.org/html/genome/ http://www.ascaa.org/ American Sickle Cell Anemia Association ASCAA was founded in 1971 and is the oldest sickle cell research, education, and social services organization in the United States. http://www.ncd.gov/ http://www.painfoundation.org/
Sites for Kids http://www.sicklecellsociety.org/sicklescene/pshomf.htm Planet Sickle Cell Society (UK based) -Youth support, Poetry, Pen-Pals, Information, Message Board http://www.starbright.org/ The STARBRIGHT Foundation is dedicated to the development of projects that empower seriousl ill children to combat the medical and emotional challenges they face on a daily basis. Coloring Books on Sickle Cell from Emory: http://www.emory.edu/PEDS/SICKLE/bbc/index.htm http://www.emory.edu/PEDS/SICKLE/chelate/index.htm
Support Group Information: Florida, Jacksonville: Sickle Cell Support Groups (904) 549-4472 Georgia, Atlanta: Parent SC Support Group (404) 616-4395