Download
1 / 32
320 likes | 581 Views
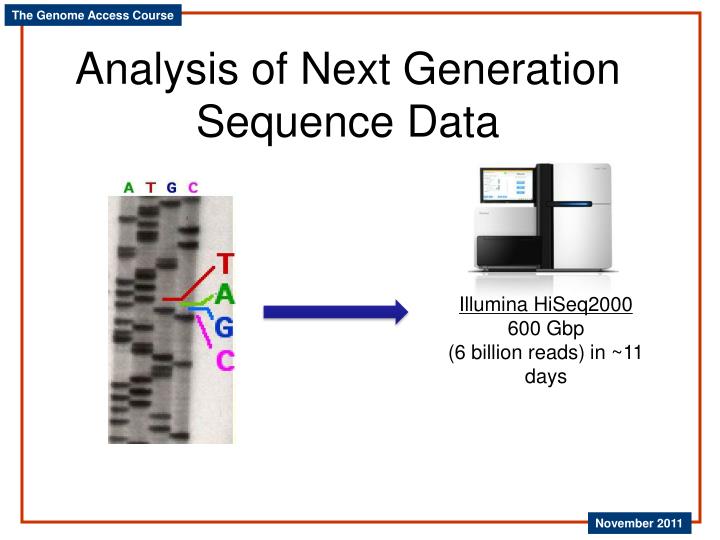
Analysis of Next Generation Sequence Data. Illumina HiSeq2000 600 Gbp (6 billion reads) in ~11 days. Typical Next Gen Experiments. Genome sequencing Novel genomes Resequencing Transcriptome sequencing (RNA-seq) Characterize transcripts with or without reference genome Typical length

E N D
Analysis of Next Generation Sequence Data Illumina HiSeq2000 600 Gbp (6 billion reads) in ~11 days
Typical Next Gen Experiments • Genome sequencing • Novel genomes • Resequencing • Transcriptome sequencing (RNA-seq) • Characterize transcripts with or without reference genome • Typical length • Short (microRNAs, …) • Find differentially expressed transcripts • Other • Methyl-seq • ChIP-seq • RIP-seq • …
Types of Sequencing Libraries Single-End Reads - 5’ or 3’ (random) Paired-End Reads - 5’ and 3’ 200-500 bp Mate-Pair Reads - 5’ and 3’ 2-5 kbp
What Does the Data Look Like?FASTQ File Format Sequence Quality (ASCII character for each base) > 80 million reads in one lane
Trim Sequences Prior To Analysis • Make sure sequencing adapters are removed • Trim ends of sequence based on quality scores
Sequence Composition Diagnostics Unbiased Reads Biased Reads First Position Nearly Always “T”
Workflows for Genome Sequencing Novel Genome Sequencing Resequencing Align reads from a sample to a reference genome assembly to examine variation BWA mapping software • de novo assembly • Generate contigs and scaffolds using overlapping reads • If applicable, align reads from a sample back to consensus to examine variation
Sequence Alignment/Map (SAM) Format • Common file format to store reads and their alignment to a reference sequence • Generated by most next gen analysis software • samtools software package
Binary Alignment/Map (BAM) Files • SAM (text file) BAM (binary file) • Not human-readable • Smaller file sizes • BAM is widely used: • Often deposited to Gene Expression Omnibus (GEO) at NCBI • UCSC Genome Browser can display alignments as a track
Glo1 CNV Present in Mouse Genomes Data for A/J Proximal Flank Chr17: 30.5Mb Max ~50x coverage Glo1 Locus Chr17: 30.7Mb Max >100x coverage Distal Flank Chr17: 31.2Mb Max ~50x coverage 50kb 50kb 50kb
Glo1 CNV Not Present in Mouse Genomes Data for NZO Proximal Flank Chr17: 30.5Mb Max ~25x coverage Glo1 Locus Chr17: 30.7Mb Max ~25x coverage Distal Flank Chr17: 31.2Mb Max ~25x coverage 50kb 50kb 50kb
RNA-Seq Reads are randomly sampled fragments from RNA sample a Proportion of reads for a transcript Expression level of transcript Garber et al, Nat Methods (2011) Lots of reads needed to construct models for every alternatively spliced transcript
Experimental Design Auer & Doerge Genetics (2010) 185: 405-416
Comparison of Affy and RNA-seq Marioni et al, Genome Res (2008) 18(9):1509-17
Comparison of Affy and RNA-seq Marioni et al, Genome Res (2008) 18(9):1509-17
Workflows for RNA-seq QC Reads Novel Transcriptome Sequencing Transcriptome Sequencing with Reference Genome Align reads from each sample/group to genome Statistics for each transcript model Examine isoforms • de novo assembly • Align reads from each sample/group to assembly • Statistics for each transcript contig Analyze Counts
de novo Transcriptome Assembly How much sequencing is enough? Rarefaction Plot
Mapping Reads Align reads to a reference Genome assembly Transcriptome assembly Commonly used aligners: bwa bowtie
RNAseq Workflow With Reference Genome Langmead et al. Genome Biology (2010), 11:R83
Map Reads & ObtainCount Reads Per Gene Both utilize a reference genome
Bowtie/TopHat Bowtie uses Burrows-Wheeler indexing for rapid mapping TopHat uses Initially Un-Mapped (IUM) reads to find novel splice sites Trapnell, Pachter, Salzberg. Bioinformatics (2009) 25(9):1105-1111
Cufflinks FPKM = Fragments Per Kilobase of transcript per Million fragments mapped Trapnell et al. Nature Biotech (2010) 28(5):511-515
Galaxy Can be used to upload FASTQ files and then run a number of QC tools and many other tools: bwa bowtie tophat cufflinks …