Download

1 / 90
1.52k likes | 2.88k Views
Vibrational Spectroscopy. and. (2) The dimension of the motion CM: With energy E, magnitude of a harmonic oscillator is: and the particle can be only found in [-A, A]. QM: Anywhere. The Comparison between a Classical Harmonic Oscillator and Its Quantum Counterpart.

E N D
and (2)The dimension of the motion CM: With energy E, magnitude of a harmonic oscillator is: and the particle can be only found in [-A, A]. QM: Anywhere The Comparison between a Classical Harmonic Oscillator and Its Quantum Counterpart According to classical mechanics, the position and energy of a harmonic oscillator are: (1) The description of the motion CM: Coordinate (x) and velocity (dx/dt) tell the exact location of the particle QM: Wave functions tell the probability at certain location.
(3)The frequency of the motion • CM: how many times the particle travels in between x = -A and x = A • QM: A simplification of analogous to the CM term. (4)Nodes and Wave Property CM: 2 nodes, no wave property QM: v nodes, wave-particle duality
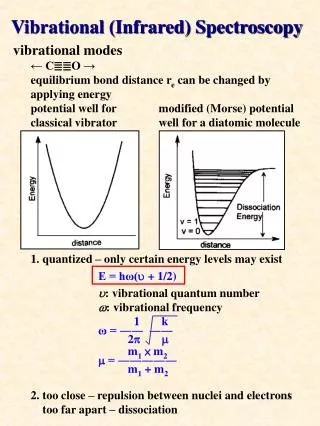
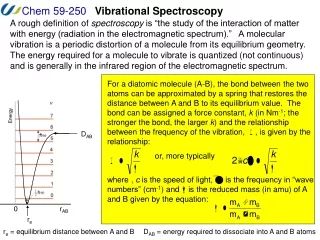
The force constant is a measure of the curvature of the potential energy close to the equilibrium extension of the bond. A strongly confining well (one with steep sides, a stiff bond) corresponds to high values of k.
A molecular potential energy curve can be approximated by a parabola near the bottom of the well. The parabolic potential leads to harmonic oscillations. At high excitation energies the parabolic approximation is poor (the true potential is less confining), and is totally wrong near the dissociation limit.
The Morse potential energy curve reproduces the general shape of a molecular potential energy curve. The corresponding Schrödinger equation can be solved, and the values of the energies obtained. The number of bound levels is finite. The illustration also shows the relation between the dissociation energy, D0, and the minimum energy, De, of a molecular potential energy curve.
Pure Vibrational Spectra of Diatomics • Harmonic Oscillator Potential • Selection Rule • Wavelengths of Transitions • The series of equally spaced energy levels gives a single line in the spectrum.
The anharmonic oscillator Real bonds, although elastic for small compressions and extensions, do not strictly obey Hooke’s Law for amplitudes > 10% of bond length. Empirical expression derived by P.M. Morse Morse Function for the potential energy: Where a is a constant for a particular molecule
When this equation is used in the Schrödinger equation, the pattern of allowed vibrational energy levels is found to be: e is always small and positive (~ +0.01) for bond stretching vibrations, so that the vibrational levels crowd more closely together with increasing u. This is only an approximation more precise expressions for the energy levels require cubic, quadratic, etc. terms in (u+1/2) with anharmonicity constants ye, ze etc. rapidly diminishing in magnitude. these values are important only at very large values of u and we shall ignore them.
Rewriting term values: And compare with the energy levels of the harmonic oscillator anharmonic oscillator behaves like the harmonic oscillator but with an oscillation frequency which decreases steadily with increasing u This is the case for any real state with v specified as a positive integer.
Ground state (u = 0) Unlike the harmonic oscillator, we cannot be measured directly. Wavenumbers for the (u +1)- u transitions are given by:
Measuring anharmonicity • We ignore the higher order anharmonicity constants. The dissociation energy is then given approximately by: • In order to determine the equilibrium frequency and anharmonicity constant, at least two transition wavenumbers must be obtained.
From the following separations of vibrational levels in the ground electronic state of CO, obtain values of we, wee , and De. Note that in practice wee is used as a single constant. It is written as a product of a constant and frequency for historical reasons. Example problem
Solution • Plotting DG vs. (2u+2) gives a straight line of slope - wee and intercept we. A least squares fit gives • we = 2171.1 ± 0.4 cm-1 • we e = 13.8 ± 0.1 cm-1
Anharmonic Oscillator Spectra • Selection Rule • For most molecules only the first three energy transitions are important. • Transitions
Important Transitions • Fundamental (first harmonic) • This transition will have strong intensity. • First overtone (second harmonic) • This is a weak intensity transition.
Not So Important Transitions • Second overtone (third harmonic) • This transition is normally too weak to be observed. • Only absorption from u = 0 need to be considered below 200 C because virtually all of the molecules are in the ground vibrational energy state.
Vibrational-Rotational Spectroscopy for Diatomics • Harmonic Oscillator Approximation • Anharmonic Oscillator
Spectrum • The vibration-rotation spectrum appears as series of lines called bands or branches, classified according to the J of the transition. • J= -2 -1 0 1 2 • Branch O P Q R S • Line Spacing • The spacing between lines is .
Lines • Lines in the R branch become closer together at higher values of J. • Lines in the P branch becomes farther apart at higher values of J. • The lines have different intensities due to different rotational populations.
There are double headed peaks due to H35Cl vs. H37Cl with an isotopic ratio of ~ 1:3 Example: HCl
Consider P and R branches in more detail • The higher energy state is for u=1, the lower energy for u=0 (this is the transition for the fundamental frequency). • To a first approximation, the transitions within the branches are separated by . • However, the rotational constant is inversely dependent on r2, and this increases with u. • Thus, .
Why Unequal Spacings? • R branch • The second term shifts lines to higher frequency with increasing J. • The third term is negative, and dependent on J2, so the lines become closer together in progressing along the series.
P branch • Here the second term is being subtracted, so the series is shifted to lower frequencies with increasing J. • The third term now spreads the lines out in progressing along the series.
Example: pure vibration spectrum of HCl - very intense absorption at 2886 cm-1 - weaker absorption at 5668 cm-1 - very weak absorption at 8347 cm-1 • To determine equilibrium frequency of the molecule – must solve any two of the 3 equations: • e (1-2e ) = 2886 cm-1 • 2e (1-3e ) = 5668 cm-1 • 3e (1-4e ) = 8347 cm-1
e = 2990 cm-1, e = 0.0174 • for the ideal harmonic oscillator the spectral absorption occurs exactly at the classical vibration frequency, but for real anharmonic molecules the observed fundamental absorption frequency and the equilibrium frequency may differ considerably. The force constant of the bond in HCl may be calculated directly from the equation when the fundamental constants and the reduced mass are inserted
The dissociation energy is the sum of the separations of the vibrational energy levels up to the dissociation limit just as the length of a ladder is the sum of the separations of its rungs. DG is the transition wavenumber
The Diatomic Vibrating – non-rigid Rotor A typical diatomic molecule has rotational energy separations of 1-10 cm-1, while vibrational energy level separations of HCl were nearly 3000 cm-1. Since the energies of the two motions are so different, as a first approximation consider that a diatomic molecule can execute rotations and vibrations quite independently (e.g., Born – Oppenheimer approximation Etotal = Eelectronic+ Evibration+ Erotation) then Etotal = Evibration+ Erotation or total = vibration+ rotation cm-1 (later will see where this approximation does not apply)
Initially ignore the small centrifugal distortion constants D, H etc., and hence: Note: retention of D has only very minor effect on the spectrum, however, it is not logical to ignore D as this implies that the molecule is rigid yet vibrating.
Selection rules for the combined motions are the same as those for each separately. • v = 1, 2, ….. J = 1 (Actually may also have v = 0, but this corresponds to the purely rotational transitions. Note, however, that a diatomic molecule, except under very special and rare circumstances, may not have J = 0, in other words a vibrational change must be accompanied by a simultaneous rotational change.) Designate rotational quantum numbers in the v = 0 state as J” and in the v = 1 state as J’ use of single prime for the upper state and double prime for the lower state is conventional in all branches of spectroscopy.
Rotational levels J” are filled to varying degrees in any molecular population, so the transitions will occur with varying intensities. If consider only the v = 0 v = 1 transition, then: Note: taking B to be identical in the upper and lower vibrational states is a direct consequence of the Born-Oppenheimer approximation – rotation is unaffected by vibrational changes.
Now we can have: (i) J = + 1 i.e. J’ = J”+1 or J’ – J” = +1 (ii) J = -1 i.e. J” = J’+1 or J’ – J” = -1 These two expressions may conveniently be combined into: Where m replaces J”+ 1 in eqn. (i) and J’+ 1 in eqn. (ii) m has a positive value for J = + 1 m has a negative value for J = -1
Note: m can not be 0, since this would imply values of J’ or J” to be -1. The frequency is usually called the band origin or band center. This equation represents the combined vibration-rotation spectrum It will consist of equally spaced lines (spacing = 2B) on each side of the band origin But since m 0, the line at itself will not appear. - Lines to the low frequency side of correspond to negative m (i.e., J = -1) are referred to as the P branch. - Lines to the high frequency side of correspond to positive m (i.e., J = + 1) are referred to as the R branch.
Inclusion of the centrifugal distortion constant D leads to the following expression for the spectrum: Note: B is 10 cm-1 or less D is 0.01% of B Since a good IR spectrometer has a resolving power of ~ 0.5 cm-1 D is negligible to a very high degree of accuracy But, the anharmonicity factor is not negligible It affects not only the position of the band origin, since But by extending the selection rules to include v = 2, 3 etc. also allows the appearance of overtone bands having identical rotational structure. From the band centers can calculate the equilibrium frequency and the anharmonicity constant
Breakdown of the Born-Oppenheimer Approximation: the interaction of rotations and vibrations for diatomic vibrating -rotator Value of B will depend on the v quantum number e.g., for v = 0 v = 1, take respective B values as B0 and B1 with B0 > B1 This gives: Where positive m values refer to the R branch and negative to P Since B0 > B1, the last term is always negative irrespective of the sign of m. The effect on the spectrum of a diatomic molecule is to crowd the rotational lines more closely together with increasing m on the R branch side, while the P branch lines become more widely spaced as (negative) m increases. Normally B1 and B0 differ only slightly and the effect is marked only for high m values.
Vibrating-Rotor for Diatomics • As a molecule vibrates the r changes and hence I changes. The rotation vibration interaction term accounts for this effect.
Combination Differences • This is a method used to determine the two rotational constants, • It involves setting up expressions for the difference in the wavenumbers of transitions to a common state; the resulting expression then depends solely on properties of the other state.
Common Upper States • The transitions have a common upper state, and hence depend on • From the equations above, it can be shown that
Obtaining Rotational Constants • A plot of the combination difference against J+1/2 should be linear with a slope of , so the rotational constant of the molecule in the state =0 can be determined. • Similarly, have a common lower state, so their combination difference can be used to determine the rotational constant of the molecule in the state =1.
Example Problem • The fundamental infrared band of H35Cl has lines at the following wavenumbers: 2775.77, 2799.00, 2821.59, 2843.63, 2865.14, 2906.25, 2925.92, 2944.99, 2963.35, 2981.05, 2998.05 cm-1. Use the method of combination differences to determine the rotational constants B0 and B1.
Procedure • Identify P and R branches, and assign J values to each line. Note that R branch (DJ = +1) starts at J = 0, but the P branch (DJ = -1) starts at J = 1. • Calculate the two series of combination differences. • Make plots and do linear regression to obtain slope = 4B.
Vibrational Spectra of Polyatomics • Selection Rules • In the harmonic approximation the vibrational quantum number may change for only one normal mode at once. • The motion must modulate the dipole moment of the molecule, i.e., it must cause the dipole moment to oscillate in value.
Not all vibrational modes are IR active • Example: CO2 • Both bends and the asymmetric stretch are active, but symmetric stretch is IR inactive. Why not? • The normal mode must modulate the molecule's dipole moment. This is not the case for a symmetric stretch of any Dh molecule.
Dipole Moments • If a molecule has a permanent dipole moment, all or most of its modes will modulate the dipole and thus be IR active. • If there is not a permanent dipole, some modes can produce a fluctuating dipole. • Determining whether a given normal mode will give rise to a vibrational band can often be done by inspection, i.e., look at the mode to see whether the motion leads to modulation of the dipole moment. • Group theory can also be used, and is sometimes necessary for complicated cases.
Each normal mode may show Q, P, and R branches • In CO2 the two bending modes can produce a motion that is the same as a rotation of a bent molecule. • This results in J = 0 (Q branch) being allowed.
Combination bands are possible • Two (or more) normal modes can change their quantum numbers at once. For a combination band each change is positive. • E.g., u = +1 for one mode u = +1 for another mode • The resultant wavenumber is about equal to the sum of the wavenumbers for the fundamental bands.
Difference bands • Concept is similar to combination bands. • One u is positive, the other is negative. • The resultant wavenumber is about equal to the difference between the fundamental bands.
Overtones • u = +2, +3 for one band. Higher overtones are possible but will be extremely weak. • The resultant wavenumber is approximately a multiple of the fundamental frequency. • As with combination and difference bands, the difference from predicted values is due to anharmonicity effects.
Example spectrum: H2O • Fundamental frequencies occur at 3756 cm-1 (asymmetric stretch), 3652 cm-1 (symmetric stretch) and 1595 cm-1 (bend). Predict other observed bands.
Self-test • Which of the following molecules have infrared active vibrations?Do not use group theory for this, only the modulating dipole rule. • H2, NO, N2O (bent), OCS (linear), CH4