Download
1 / 43
480 likes | 820 Views
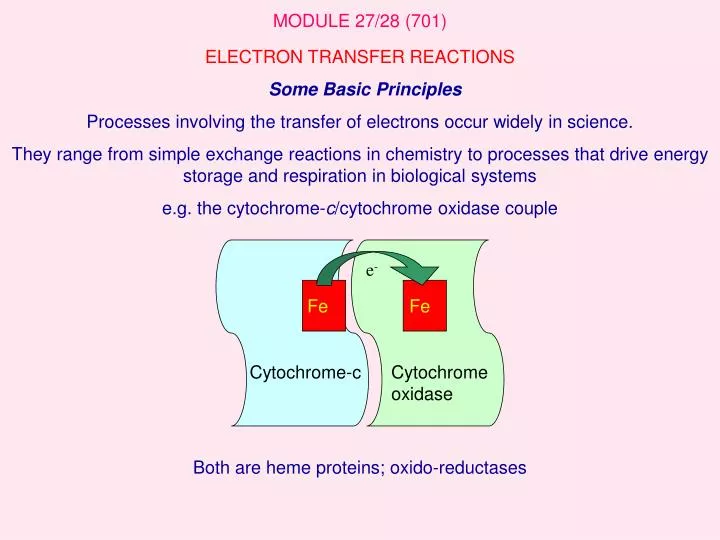
Fe. Fe. e -. Cytochrome-c. Cytochrome oxidase. MODULE 27/28 (701). ELECTRON TRANSFER REACTIONS Some Basic Principles Processes involving the transfer of electrons occur widely in science.

E N D
Fe Fe e- Cytochrome-c Cytochrome oxidase MODULE 27/28 (701) ELECTRON TRANSFER REACTIONS Some Basic Principles Processes involving the transfer of electrons occur widely in science. They range from simple exchange reactions in chemistry to processes that drive energy storage and respiration in biological systems e.g. the cytochrome-c/cytochrome oxidase couple Both are heme proteins; oxido-reductases
MODULE 27/28 (701) • Photoscientists are keenly interested in electron transfer reactions because: • they occur as primary events in many photoprocesses. • they can be conveniently studied using photophysical techniques. • photophysical methods offer excellent ways of testing the theories. Kinetic Aspects of Bimolecular Reactions An electron transfer reaction between individual molecules freely diffusing in a mobile liquid has characteristics of all such bimolecular reactions: The reactants diffuse together, react, and two different entities diffuse apart. The process proceeds via a collision complex, one or more reaction intermediates, or a transition state. Micro-reversibility applies, and the process occurs on a continuous potential energy surface. No excited states have yet been invoked, so we can imagine the process to be adiabatic (no crossings to other PE surfaces) — not always true.
MODULE 27/28 (701) We can define rate constants for both forward and reverse processes, kf and kr, These need to be measured and the factors that influence them understood. The overall reaction, like all chemical reactions, will have characteristic Keq,DG0, DH0, and DS0parameters. In addition, we can relate DG0 to reduction potentials, viz., Thermodynamic constants are useful for describing equilibrium states-- no information on mechanistic details. Experiment shows that for exoergic electron transfer processes, rate constant values occur over a wide range, up to the limit imposed by diffusion. kobs = f.kd This “f” needs to be understood. Consider a detailed scheme for the overall bimolecular process shown above.
D + A D+ + A- kd k-d k-d kd kn ne k’-n k-n ne k’n [DA] Precursor complex [DA]# Reorganized precursor complex [D+A-]# Successor complex [D+A-] Reorganized successor complex MODULE 27/28 (701)
MODULE 27/28 (701) • A steady state treatment leads to • Where if • When ka >> kd, then kobs kd • Let us examine the expression for ka • Under conditions when ne > k-n i.e., the electron moves to A more • rapidly than the reorganized complex relaxes, then • Here k-n is the rate of relaxation of vibrationally excited precursor, and DG#n is the energy barrier to nuclear reorganization. • Under these conditions ka is independent of ne (the electron-hopping rate).
MODULE 27/28 (701) (ii) On the other hand, whenk-n > ne In both cases (and all others), the nuclear reorganization process is a barrier to electron transfer and imposes an activation step. Thus the overall (measured) rate constant is a combination of diffusion-dependent (kd) and activation-dependent (ka) terms: Therefore when ka >> kd, then kobs kd. and our kinetic measurements can provide no information on the activation-dependent process because everything is limited by diffusion.
MODULE 27/28 (701) However, the central segment of the sequence occurs independently of how the sequence is initiated. To investigate the role of activation we need to circumvent the diffusion limiting problem. Later we will see how this can be done in a practical way. For now, we assume that it can be done and proceed to examine ka. The parameters k-n, ne, and DGn# are important in determining the magnitude of ka. Theoreticians have examined these using classical mechanics Marcus, Sutin, Hush And semi-classical/quantum methods Jortner, Levich.
MODULE 27/28 (701) THE TRANSITION STATE APPROACH Our scheme is The steps prior to (D/A) and those after electron transfer are ignored. D and A may be polyatomic molecules, aquated metal ions, etc., and the reaction above proceeds with changes in bonding coordinates in D and A, and solvation around the complex. (D/A)# and (D+/A-)# have identical nuclear configurations, differing only in that a single electron has switched its molecular assignment. The situation resembles a Franck-Condon type event, or a radiationless transition between two states. Using the radiationless transition approach, we write where r is an average density of states in the acceptor.
MODULE 27/28 (701) In electron transfer theory, r usually appears as a Franck-Condon weighted density of states (FCWD). The electronic matrix element term be2 contains the operator driving the process. The classical picture due to Marcus (1956) is less rigorous and simpler, but provides useful physical insights. It provides a comparison to the transition state theory (TST) of kinetics. Marcus chose to represent the complex multidimensional PE surfaces of polyatomic reactant pairs as a parabolic energy curve in "nuclear configuration space"
MODULE 27/28 (701) REACTANT STATE PRODUCT STATE DG l DGn# Nuclear configuration • is the energy required to move the electron in the (D/A) to (D+/A-) without prior nuclear reorganization (resembles a Franck-Condon event).
MODULE 27/28 (701) DGn# is the energy required to reconfigure the precursor complex to a non-equilibrium nuclear configuration in which the electron transfer can occur The system switches from the reactant state surface to the product state surface. Note thatDGn# < l At the curve crossing, the electron can hop from one curve to another with some probability (rate). The situation shown in the schematic is for DG0= 0 an isoergic process. Marcus recognized he could analyze the situation using the analytical geometry of intersecting parabolas WhenDG0≠ 0 Thus the energy barrier (DGn#) to electron transfer depends on DG0 and l in a quadratic manner.
MODULE 27/28 (701) DG0 The effect on DG#n as the value of DG0 becomes increasingly negative: As the product parabola is lowered wrt the reactant curve, the nuclear reorganization barrier first becomes less and then increases again the only change is in the overall driving force, DG0 no shape changes, no shifts in curve minimum.
MODULE 27/28 (701) inverted region normal region ka l Earlier we saw that with or Thus, Marcus theory predicts that for weakly exoergic reactions that log ka increases as -DG0 increases. It maximizes at and it decreases again as -DG0 increases beyond l (invertedregion) This remarkable result flies in the face of intuition — WHY? It led to the Nobel Prize in Chemistry for Rudy Marcus in the early 1990s
MODULE 27/28 (701) Nuclear coordinate We have assumed that the product is formed in its zeroth vibrational state. This is a simplification and in fact the formation of product species in vibrational states above the zero point is very possible. DG The dashed curves represent four vibrational states of the product. The red dashed curve (v = 0) crosses the reactant curve in the inverted region, the higher vibrational modes do not.
MODULE 27/28 (701) The rate constant observed will be a weighted sum of the contributions from all the modes. It will be larger than that if the v = 0 mode was the only contributor. Thus the inverted region will be less pronounced than otherwise; the parabola will depart from the symmetrical form
MODULE 27/28 (701) The Marcus approach, being geometrical, assumes symmetrical sets of pure parabolas. Which means that there is only very weak interaction between surfaces at the crossing point. Thus the reaction is by necessity non-adiabatic, since curve crossing must occur.
MODULE 27/28 (701) At the crossing point the two states are degenerate. Time dependent perturbation theory tells that the probability of transferring to the product curve will be proportional to The vibrational motion of the nuclei is such that the system spends only a short time in the crossing region. For states where lVlis small (< kBT), the probability of reaction will be small, and the system will continue on the reactant surface for many passages through the crossing region. When the perturbation is large (> kBT) (adiabatic) the argument of the sine is large. The oscillatory frequency will be sufficient to ensure effective passage into product space.
REACTANT PRODUCT DG l DGn# Nuclear configuration MODULE 27/28 (701) The Reorganization Energy The barrier to electron transfer, per Marcus, is manifest as a free energy term composed of DG0 and lcomponents. The former is a thermodynamic state property, defining the overall free energy changes in going from reactants to products. The quantity lis an energy term that is identified by Marcus as the energy of a FC transition from the relaxed precursor state into the nuclear configuration space of the product state.
MODULE 27/28 (701) lhas two additive parts: • l0 - inner shell, derived from required changes in the internal nuclear geometry of the donor and acceptor molecules. • ls - outer shell, arising from the required readjustment of solvent dipoles to accommodate the shift in the electronic charge. • Marcus (1959), assuming that the solvent behaves as dielectric continuum (no local structure; hard sphere molecules) derived the following expression:
MODULE 27/28 (701) For polar organic liquids (CH3CN) ls ~ 0.75 V For non-polar organic liquids (cyclohexane) ls ~ 0.15 V The value of lo is not easily calculated. It is usually estimated from considerations of the force constants of normal mode vibrations in the reactant and product species. A typical value for lo is ~ 0.4 V For some exchange reactions such as (H2O)6Fe2+/3+ and (NH3)6Co2+/3+, the redox change necessitates large M ligand bond length changes (140 and 220 pm respectively). These lead to lo values of 8.4 and 17.6 kcal mole-1 respectively.
MODULE 27/28 (701) The ne parameter ne has been used to represent the frequency (or rate) at which an electron can shift between (D/A)# and (D+/A-)# . This can be expanded as nn is the frequency for nuclear reorganization and Kel is the probability of an electron transferring from one PE curve to another in the reorganized configuration. In TST terms, nn can be regarded as being similar to an entropic term, viz., The maximum value (DS# = 0) of nn is kBT/h, approx 1013 s-1
MODULE 27/28 (701) Kel is similar to the transmission coefficient of TST. It takes values between zero and one. It represents the probability of the system moving from reactant to product surface. it depends on the interaction energy between the two surfaces at the crossing region. V is the electronic coupling matrix element; equivalent to beused earlier.
MODULE 27/28 (701) D RDA A D RDA A Molecular wave functions decrease exponentially with distance from their maximum amplitude. Overlap increases as the distance between the reacting species decreases.
MODULE 27/28 (701) • The interaction potential between the MOs is propnl to exp(-brRDA) where br (not to be confused with be ) is a multiplier of RDA having dimensions of m-1 • It is equivalent to a molecularresistance to electron transfer. • If br is small for a given RDA , then the interaction is large and electron transfer occurs effectively. • if br is large for a given RDA , then the interaction is less and the transfer efficiency is reduced. • Thus the overall rate constant for electron transfer in the activated case (ka) depends on • distance between the participants • the frequency with which nuclei reorganize • a reorganization barrier
MODULE 27/28 (701) Overall the activated rate constant can be described by The maximum value of nn is approximately 1013 s-1. The electronic coupling factor, Kel, depends on RDAand on the relative orientation between the dipoles in the donor and acceptor. The l parameter is the reorganization energy, which depends on solvent reorientation and on nuclear changes internal to the molecules.
MODULE 27/28 (701) Our development has been independent of the electronic state of the reacting species. Electron Transfer in the Photosciences Advantages of photoexcitation 1No transfer occurs until the photons are absorbed. Systems can be manipulated (synthesized, mixed, etc.) in ground states. 2Wavelength specificity allows preferential excitation of one component. 3 Absorption is virtually instantaneous zero "dead" time. 4Two rate constants can often be determined for each D/A couple (Figure over). 5Electronically excited states have > 1 eV more energy than the parent ground states and are therefore better oxidants and reductants.
MODULE 27/28 (701) D* + A kET D+ + A- k-ET D + A
MODULE 27/28 (701) • Experimental Observations: • Measurements of bimolecular electron transfer rate constants permeate the literature, but they are of little use in testing the theoretical picture since • distance and orientation effects are averaged over the population of reactants. • diffusion effects tend to obscure details of thelog kvs. DG0plot at highDG0. • (recall the discussion on diffusion-controlled bimolecular processes) 0
MODULE 27/28 (701) log k diffusion limit 0 increasing driving force DG0 As the driving force increases, so does the bimolecular rate constant for the reaction (the normal region). However at some point, even though the driving force continues to increase, the rate constant levels off because the rate-limiting step is now the diffusion of the reactants together. Rehm-Weller plots
MODULE 27/28 (701) Progress made by confining D and A in close proximity (no need for diffusion to bring reactants together). Three basic approaches to this: 1: Retain D and A as discrete molecules, but disperse them in rigid glasses (J.R. Miller, 1975; G. McLendon, 1983). A relatively crude approach since a distribution of RDA values generated.b ~ 0.01 pm-1. 2: Link D to A via a rigid spacer, using covalent bonding Variations in spacer length allow RDA changes to be effected (keep DG0constant) Change E0D+/D and EA/A- at constant RDA allows DG0 effects to be investigated. (Closs and Miller seminal paper in 1983).
D A SPACER MODULE 27/28 (701) Closs and Miller employed steroids, decalins, and cyclohexanes as rigid spacers. Verhoeven and Padden-Row used a series of linked norbornanes Isied, and independently Klapper, employed oligoprolines of variable lengths Grayet al. covalently attached Ru(II) complexes to histidine residues on the surface of cytochrome-c.
MODULE 27/28 (701) MTHF ETHER iso-OCTANE The figure shows some of the Closs and Miller results The inverted region is clearly apparent in all solvents the l value shifts to lower energies as the polarity of the solvent is reduced (inner sphere contribution is lessening)
MODULE 27/28 (701) Distance effects Closs and Miller used rigid spacers of different lengths (J. Phys. Chem. (1986), 90, 3673), in a pulse radiolysis experiment, both entities pick up electrons (only one per molecule). Np- transfers electron to the Bp moiety. The measured rate constants increased as the through-bond distance decreased.
MODULE 27/28 (701) Oligoprolines form rigid bridges Klapper et allinked tyr to one end and trp to other and generated a trp radical and measured the rate parameter as this decayed to generate the tyr radical. Carrying out the experiment as function of the number of prolines in the linker led to an evaluation of b. Isied et al. linked different transition metal complexes to each end of oligopro bridges of different length and obtained distance effects. Schanze & Sauer linked Ru(II) polypyridyl complexes at one end and quinones at the other, again finding distance effects.
MODULE 27/28 (701) Protein-based systems Proteins can be modified at surface sites. The Isied and the Gray groups did this in 1982. Both groups found that Another approach is to employ electrostatically joined redox protein pairs. The Brian Hoffman (1983) and George McLendon (1984) groups showed that electron transfer can occur over distances of up to 2 nm This approach uses self-assembly of D-A systems through electrostatic interactions, and it offers a convenient way of preparing D-A couples with minimal synthetic demands.
MODULE 27/28 (701) heme Our own studies (Zhou and Rodgers, 1990) in this arena have employed this approach. Self-assembled systems composed of an anionic metalloporphyrin and cytochrome-c, which has a cationic (lysine) surface patch in the vicinity of the heme pocket.
MODULE 27/28 (701) The Fe atom in the heme can be replaced by two H atoms (free base), or by metals such as Zn or Mn. This leads to E0M/M- variation at the heme site. Uroporphyrin has a total of eight carboxylate residues on the periphery association with the lysines around the heme cleft. Similarly, the free base protons in uroporphyrin can be replaced by Zn (II), Fe(II), Mn(II)… Again this leads to E0M/M- changes in the porphyrin.
protein Up Fe heme M MODULE 27/28 (701) The ion pair is envisaged to be structured as shown Computer modeling showed the heme edge to uroporphyrin center to be 0.8 nm. The uroporphyrin/cyt-c system allows measurement of diffusion-free electron transfer at fixed distance and with variable driving force Can test the exponential term in the relationship
CIII:Up(S1) CIII:Up(T1) 2.2 eV CII:Up+ 1.6 eV 0.6 eV CIII: Up(S0) MODULE 27/28 (701) In aqueous solution at pH = 7.4, and low ionic strength the equilibrium has Keq = 106M-1; thus at [Up] = 35 mM and [cyt-c] = 65 mM, the porphyrin was 96% in form of complex. The reduction potentials for the reactants are such that when Up(T1) is formed the reduction of the cyt-c(III) is now 1 eV downhill, i.e., the photon provides the driving force for the electron transfer
MODULE 27/28 (701) 3[Up:CIII]* kCS 3[Up+: CII] kCR [Up:CIII] Cyto-c (III) with ZnUp triplet: growth and decay of separated radical pair 550 nm This experiment yields forward and reverse rate constants for electron transfer
l = 0.7 V MODULE 27/28 (701) Forward and reverse rate constants were independent of cytochrome-c concentration (required for intra-complex reactions). Plots of log k against DE for a series of donors and acceptors are shown for the thermal back reaction and the photo-induced forward reaction.
MODULE 27/28 (701) The thermal back transfer shows Marcus behavior (inverted region > 0.7 V) The reaction between the excited state and its reaction partner shows Rehm-Weller behavior (normal region only) This is most likely because the forward reaction requires significant local diffusion to occur within the complex before the required configuration is reached (gating) This would require significant readjustments of the solvent sheaths of the carboxylate and ammonium residues at the ion pairing sites, which, in turn would increase the outer sphere contribution to l. The reverse reaction does not have this requirement since the reaction partners are now in their optimal positions, and the outer sphere contribution to l is thereby lower.