Download

1 / 38
380 likes | 486 Views
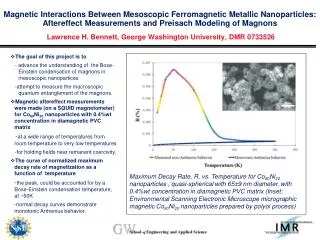
In Bioinformatics use a computational method - Dynamic Programming to align two proteins or nucleic acids

E N D
In Bioinformatics use a computational method - Dynamic Programming to align two proteins or nucleic acids The term dynamic programming to describe the process of solving problems where one needs to find the best decisions one after another. At first, we select the best path from Start to A, then we select the best path from A to Finish. The choice of the best path from A to Finish is independent of the choice of path from Start to A
Thus the path is subdivided into a set of steps. The goal is to find the optimal way for each step Any step along the true optimal path must itself be the optimal path. This is the main idea of dynamic programming method. Dynamic programming is typically used when a problem has many possible solutions and an optimal one needs to be found.
sequence 1 : S D V – Y sequence 2 : S R V L Y 2 -1 2-2 2 Sum of residues pair scores minus . gap penalty = -2 Total score =3 Score of new Score of previous + Score of new aligned alignment alignment pair sequence 1 : S D V – Y T sequence 2 : S R V L Y T Score = 5 3 + 2 .
There are two Sequences: A = ACGCTG, B = CATGT The best alignment ? Question: explain the cell in the first row and the first column
– A C G... – C A T...
Question: How do we calculate the score of this alignment?
Question: How do we estimate the mismatch? 0, -1, 1?
Question: How do we estimate the match? 0, 1, 2 Thus in this alignment the penalty for a gap is… . the score for a mismatch is…
Explain the score in the cell G3/ C1 Check the score for mismatch with the previous slides.
After filling in all of the values the score matrix is as follows:
The next procedure is the traceback step. The traceback step determines the actual alignment that result in the maximum score. The traceback step begins in the N,M position in the matrix, i.e. the position where both sequences are globally aligned
The algorithm of the traceback: a) step begins with the last cell Traceback takes the current cell and looks to the neighbor cells that could be direct predacessors: to the neighbor to the left (gap in sequence #2), the diagonal neighbor (match/mismatch), and the neighbor above it (gap in sequence #1). there is a G6/T5 in this case).
For the current cell there are two possible predacessors with the maximum score 3. b) If more than one possible predacessor ( left and above) with the same maximum score exists, any can be chosen. If the diagonal neighbor has the same maximum score, diagonal way is selected to avoid a gap. Variant 1: select left cell as the predacessor. …TG …T - Select the best alignment and compare with the alignment at the next slide.
Question: Does your alignment coincide with this one? Make another possible alignment (Variant 2) and then compare it with the alignment at the next slide.
Variant 2 Question: What are the maximum scores of these two possible alignments?
Proteins Nucleotides Codons What to align?
When are sequences Similar ? Apart from sequence similarity, it depends on: • Nucleotide / protein • Nucleotide: 4 different residues → more likely to be similar by chance • Proteins: 20 different residues → less likely to be similar by change • Sequence length • Short sequences → similarity by chance
Similarity vs. identity • Identity: the same residue • Similarity: similar physiochemical characteristics (can more readily be substituted)
Algorithms - overview • Goal: comparing conserved regions • Two methods: • Global • Local • Three techniques • Dot plots • Dynamic programming • Word-based
Local vs. global alignments sequence 1: ACTCCGTAGGTTGGACTCC sequence 2: CTCTGGTAGGCTTACTCTG Global alignment Local alignment
Agenda • Sum of Pairs method • ClustalW • Gap penalties
Sum of Pairs (SP) method • Consider aligning the following 4 protein sequences • S1 = AQPILLLV • S2 = ALRLL • S3 = AKILLL • S4 = CPPVLILV • Which MSA to choose? • A Q P I L L L V • A L R - L L - - • A K - I L L L - • C P P V L I L V A Q P I L L L V A - - L R L L - - A K I L L L - C P P V L I L V
Sum of Pairs cont. • Assume: c(match) = 1 , • c(mismatch) = -1 , • c(gap) = -2, • c(-, -) = 0. • Then the SP score for the 4th column of the MSA would be SP(column4) = SP(I,-,I,V) • = c(I,-) + c(I,I) + c(I,V) + c(-,I) + c(-,V) + c(I,V) • = -2 + 1 + (-1) + (-2) + (-2) +(-1) • = -7 A Q P I L L L V A L R - L L - - A K - I L L L - C P P V L I L V
Sum of Pairs cont. • To find SP(MSA) we would find the score of each column miand then SUM all SP(mi) scores to get the score MSA. • To find the optimal score using this method we need to consider all possible MSA.
The ClustalW method • ClustalW is a progressive method for MSA • “Progressive” • Start: pairwise determine the most related sequences • progressively add less related sequences or groups of sequences to the initial alignment.
ClustalW steps • Multiple Alignment Step: • Aligning S1 and S3 • Aligning S2 and S4 • Aligning (S1, S3) with (S2,S4) All Pairwise Alignments Dendrogram Similarity Matrix Cluster Analysis From Higgins(1991) and Thompson(1994).
ClustalW Step 1 • Use a pairwise alignment method to compute all pairwise alignments amongst the sequences. • Look at the non-gapped position and count the number of mismatches between the two sequences, then divide this value by the number of non-gapped pairs to calculate the distance • NKL-ONdistance = 1/4 = 0.25 • -MLNON
ClustalW Step 1 continued Seq. S1 S2 S3 S4 S1 - S2 0.17 - (SYMMETRIC) S3 0.59 0.60 - S4 0.59 0.59 0.13 -
S 1 S 3 S 2 S 4 ClustalW Step 2 • Construct a similarity tree (Guide tree). • The root is placed a the midpoint of the longest chain of consecutive edges. S3 S4 S1 S2
ClustalW Step 3 • Combine alignments: • from the most closely related groups to the most distantly related groups • going from tip of tree to the root of the tree. • In our example, we align: • S1 with S2 (grp1) • S3 with S4 (grp2) • grp1 with grp2 • continue until the root is reached. • Each alignment involves dynamic programming by the SP score method. • Now the complexity has been reduced to that of a series of pairwise alignments
Distance between sequences - measure from the guide tree - determines which matrix to use • 80-100% seq-id -> Blosum80 • 60-80% seq-id -> Blosum60 • 30-60% seq-id -> Blosum45 • 0-30% seq-id -> Blosum30
Gap penalties Gap Opening Penalty (GOP) Gap extension penalty (GEP) GTEAIVLMANKL G---------KL Gap Penalty: GOP+8*GEP
Modifications of gap penalty • Gap at position • low GOP (Residue specific penalties) • gap within 8 residues? -> increase GOP (Gap separation distance) • Hydrophilic residues • lower GOP (Hydrophilic-Hydrophobic gap penalties)