Download

1 / 55
740 likes | 1.29k Views
RNA structure analysis. Jurgen Mourik & Richard Vogelaars Utrecht University. Overview. Introduction to RNA RNA secondary structure prediction Nussinov folding algorithm Zuker folding algorithm Demonstration Questions. Introduction to RNA (1). R ibo n ucleic a cid To many people:

E N D
RNA structure analysis Jurgen Mourik & Richard Vogelaars Utrecht University
Overview • Introduction to RNA • RNA secondary structure prediction • Nussinov folding algorithm • Zuker folding algorithm • Demonstration • Questions RNA structure analysis
Introduction to RNA (1) • Ribonucleic acid • To many people: • “RNA is the passive intermediary messenger between DNA genes and the protein translation machinery” • But: • Many non-coding RNAs exist • Adopt sophisticated 3D structures • Catalyse biochemical reactions RNA structure analysis
Introduction to RNA (2) • Three major types of RNA • Messenger RNA (mRNA) • Serving as a temporary copy of genes that is used as a template for protein synthesis. • Transfer RNA (tRNA) • Functioning as adaptor molecules that decode the genetic code. • Ribosomal RNA (rRNA) • Catalyzing the synthesis of proteins. RNA structure analysis
RNA world hypothesis • RNA is the only biological polymer that serves as both a catalyst (like proteins) and as information storage (like DNA). • For this reason some people think that a RNA-like molecule was the basis of life early in evolution. RNA structure analysis
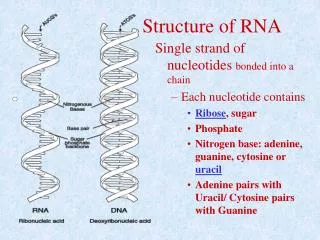
Terminology of RNA (1) • Four nucleotides: • Adenine • Cytosine • Guanine • Uracil • Canonical base pairs: • G-C • A-U • Non-canonical base pairs • G-U RNA structure analysis
Terminology of RNA (2) • Base pairs are approximately coplanar and almost always stacked onto other base pairs in a RNA structure • Contiguous stacked base pairs are called stems • In 3D, RNA stems generally form a regular double helix RNA structure analysis
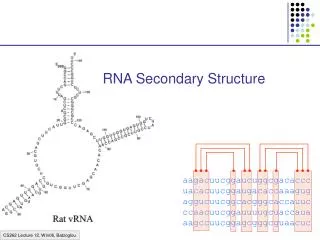
RNA secondary structure • Unlike DNA, RNA is typically produced as a single stranded molecule which then folds intramolecularly to form a number of short base-paired stems. This base-paired structure is called the secondary structure of the RNA. RNA structure analysis
Elements of a RNA secondary structure (1) • Loop: single stranded subsequence bounded by base pairs • Hairpin loop: a loop at the end of a stem • Bulge (loop): single stranded bases occurring within a stem • Interior loop: single stranded bases interrupting both sides of a stem • Multi-branched loop: a loop from which three or more stems radiate RNA structure analysis
Elements of a RNA secondary structure (2) etc. C U C G C U G ● C G ● C U ● A A ● U C ● G G G 3’ G A 5’ RNA structure analysis
Pseudoknots (1) • Base pairs almost always occur in a nested fashion in RNA secondary structure • A base pair between position i and j and a base pair between i’ and j’ are nested if and only if: • Non-nested base pairs are called pseudoknots RNA structure analysis
Pseudoknots (2) • None of the dynamic programming algorithms can deal with pseudoknots, including the Zuker and Nussinov RNA folding algorithms. • Pseudoknots occur in many important RNA’s: • The algorithms ignore biologically important information. • For database searching for RNA homologues, it is acceptable to sacrifice the information in pseudoknots. RNA structure analysis
RNA sequence evolution • The sequence evolution of RNA is constrained by the structure. • It is possible to have two different RNA sequences with the same secondary structure. • Drastic changes in sequence can often be tolerated as long as compensatory mutations maintain base-pairing complementarity. RNA structure analysis
{C,U} {A, C, G, U} Complement of base N {A,G} RNA sequence evolution (2) • Suppose we want to search for a nucleotide sequence for occurrences of consensus R17 coat protein: • It is useless to use standard sequence alignment • R17 coat protein binds and represses translation of its replicase: • It blinds most of the primary sequence positions RNA structure analysis
RNA sequence evolution (3) • How to solve this problem? • RNA pattern-matching program (RNAMOT). • Searches for deterministic (non-stochastic) motifs but with secondary structure constraints as extra terms. • Works fine for small, well-defined patterns but is somewhat insensitive and problematic for finding matches to less well conserved structures. RNA structure analysis
Inferring structure by comparative sequence analysis • In a structurally correct multiple alignment of RNAs, conserved base pairs are often revealed by the presence of frequent correlated compensatory mutations • RNA secondary prediction method: comparative sequence analysis • The accepted consensus structures of most well-studied RNAs have been derived by comparative analysis. RNA structure analysis
Problem! How does comparative sequence analysis work? (1) • Inferring the correct structure by comparative analysis requires knowing a structurally correct alignment • Inferring a structurally correct multiple alignment requires knowing the correct structure RNA structure analysis
How does comparative sequence analysis work? (2) • Solution: make use of an iterative refinement process of: • Guessing the structure based on the current best guess of the alignment • Realigning based on the new guess at the structure • The sequences to be compared must be: • Sufficiently similar to start the process • Sufficiently dissimilar that a number of co-varying substitutions can be detected RNA structure analysis
The frequency of one of the four bases observed in column i. Mijvaries between 0 and 2 bits The joint (pairwise) frequency of one of the sixteen possible base pairs observed in columns i, j. Mutual information (1) • A quantitative measure of pairwise sequence covariation • Given two aligned columns i, j, the mutual information is given by: RNA structure analysis
Mutual information (2) • Mij tells us how much information we get about the identity of the residue in one position if we are told the identity of the residue in the other position • If you know that i is a G, the uncertainty about j collapses from four different possibilities to just one (C) 2 bits of information • If i and j are uncorrelated, the mutual information is zero RNA structure analysis
RNA secondary structure prediction (1) • Many plausible secondary structures can be drawn for a sequence • But: the number of secondary structures increases exponentially with sequence length • An RNA of 200 bases has over 1050 possible base-paired structures • Goal: distinguish the biologically correct structure from all the incorrect structures. RNA structure analysis
RNA secondary structure prediction (2) • We need: • A function that assigns the correct structure the highest score • An algorithm for evaluating the scores of all possible structures • Two methods: • Nussinov folding algorithm • Zuker folding algorithm RNA structure analysis
Need a break? Well here it is!
Nussinov folding algorithm (1) • Goal: Find the structure with the most base pairs • Nussinov introduced an efficient dynamic programming algorithm for this problem • A recursive algorithm that calculates • the best structure for small subsequences and • works its way outwards to larger and larger subsequences RNA structure analysis
Nussinov folding algorithm (2) • Key idea of recursion: • There are only four possible ways of getting the best structure for i,j from the best structure of the smaller subsequences • Two stages: • Fill stage of the algorithm • Trace back stage of the algorithm RNA structure analysis
Nussinov folding algorithm (3) • The four possible ways: • Add unpaired position i onto the best structure for subsequence i+1,j • Add unpaired position j onto the best structure for subsequence i,j-1 • Add i,j pair onto best structure found for subsequence i+1,j-1 • Combine two optimal substructures i,k and k+1,j RNA structure analysis
Nussinov folding algorithm (4) • Formal description of the algorithm: • Given a sequence x of length L with symbols xi,…,xL • Let if xiand xj are complementary base pairs else • Recursively calculate scores which are the maximum number of base pairs that can be formed for subsequence xi,…,xL RNA structure analysis
Nussinov algorithm: fill stage • Initialisation: • Recursion: starting with all sub sequences of length 2, to length L: RNA structure analysis
Example sequence: GGGAAAUCC RNA structure analysis
A*U= base pair Example sequence: GGGAAAUCC RNA structure analysis
This value gives the maximum nr. of base pairs Example sequence: GGGAAAUCC RNA structure analysis
Nussinov algorithm: traceback stage • Initialisation: Push (1,L) onto the stack. • Recursion: Repeat until stack is empty: RNA structure analysis
Initialisation: Push (1,L) Example sequence: GGGAAAUCC RNA structure analysis
Recursion: Example sequence: GGGAAAUCC RNA structure analysis
Example sequence: GGGAAAUCC RNA structure analysis
A A A ● U G ● C G ● C G Example sequence: GGGAAAUCC RNA structure analysis
SCFG version of the Nussinov algorithm • Stochastic Context-Free Grammars • Will be discussed next Wednesday • Makes use of production rules: • S aS | cS | gS | uS (i unpaired) • Every production rule has a associated probability parameter. • The maximum probability parse is equivalent to the maximum probability secondary structure. RNA structure analysis
Chomsky normal form: • All context free grammar production rules are of the form: • S SS or • S a Needed terminology • The inside-outside (recursive dynamic programming) algorithm for SCTGs in Chomsky normal form is the natural counterpart of the forward-backward algorithm for HMM. • Best path variant of the inside-outside algorithm is the Cocke-Younger-Kasami (CYK) algorithm. It finds the maximum probabilistic alignment of the SCFG to the sequence. Just as the viterbi algorithm for HMMs RNA structure analysis
CYK for Nussinov-style RNA SCFG (2) • Initialisation: • Recursion: Addition to the fill stage of the Nussinov algorithm. The principal difference is that the SCFG description is a probabilistic model. RNA structure analysis
CYK for Nussinov-style RNA SCFG (2) • The is the log likelihood of the optimal structure given the SCFG model • The traceback to find the secondary structure corresponding to the best score is performed analogously to the traceback in the Nussinov algorithm RNA structure analysis
CYK for Nussinov-style RNA SCFG (3) • Good starting example (10.2), but it is too simple to be an accurate RNA folder • The algorithm does not consider important structural features like preferences for certain: • Loop lengths • Nearest neighbours in the structure caused by stacking interactions between neighbouring base pairs in a stem. RNA structure analysis
Zuker folding algorithm (1) • Most sophisticated secondary structure prediction method for single RNAs • An energy minimisation algorithm which assumes that the correct structure is the one with the lowest equilibrium free energy • The of an RNA secondary structure is approximated as the sum of individual contributions from loops, base pairs and other secondary structure elements. RNA structure analysis
Zuker folding algorithm (2) • Difference with the Nussinov folding algorithm: • Energies of stems are calculated by adding stacking contributions for the interface between neighbouring base pairs instead of individual contributions for each pair. • Advantage: • Better fit to experimentally observed values for RNA structures, but it complicates the dynamic programming algorithm RNA structure analysis
Zuker folding algorithm (3)Freier energy rules • The energies in the tables are from the older ‘Freier rules’ at 37ºC. • For more information see the article ”Improved free-energy parameters for predictions of RNA duplex stability” by Freier et al. RNA structure analysis
Zuker folding algorithm (4) RNA structure analysis
Zuker folding algorithm (5) RNA structure analysis
Zuker folding algorithm (6) • The minimum energy structure can be calculated recursively by a dynamic programming algorithm very similar to how the maximum base-paired structure was calculated like the Nussinov algorithm. • Now we keep two matrices • W(i,j) is the energy of the best structure on i,j • V(i,j) is the energy of the best structure on i,j given that i,j are paired. RNA structure analysis
Suboptimal RNA folding(CYK algorithm will be explained next Wednesday) • The original Zuker algorithm finds only the optimal structure. • The biologically correct structure is often not the calculated optimal structure. • Zuker introduced a suboptimal folding algorithm. • Is similar to running the CYK algorithm in both inside and outside directions. • The algorithm samples one base pair sub optimally. • The rest of the structure is the optimal structure given that base pair. RNA structure analysis
Demonstration RNAstructure By David H. Mathews Michael Zuker Doulas H. Turner
Demo: RNAstructure (1) • The core of RNAstructure is a dynamic programming algorithm to predict RNA or DNA secondary structures from sequence based on the principle of minimizing free energy. RNA structure analysis