Download

1 / 2

E N D
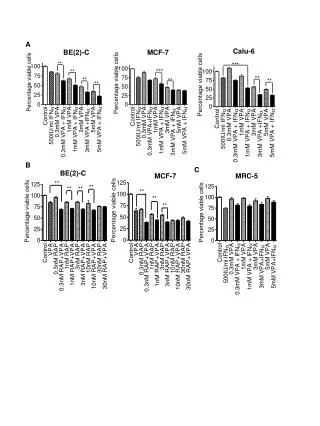
鎘誘導人類正常肺細胞MRC-5細胞凋亡之探討 • 鎘 (cadmium)為一環境污染物,其半衰期長達10-30年,主要經由香菸、食物及空氣污染進入人體,可導致肺臟、腎臟、肌肉、血管與神經退化性疾病。鎘引起細胞凋亡 (apoptosis)之訊息傳遞路徑尚未完全明瞭,本論文以人類正常肺纖維母細胞MRC-5為細胞模式,探討鎘傷害細胞的機制。以AnnexinV/PI雙染色,檢測鎘濃度(25-150 mM)與細胞死亡之關連性,顯示100 M鎘可誘導40%細胞凋亡及20%壞死,其細胞凋亡比例達高原期 (plateau),並隨鎘濃度增加而減少,但細胞壞死比例略增,因此選用鎘100 mM,進行後續實驗。廣泛性caspase抑制劑 (Z-VAD.fmk)無法防止細胞走向凋亡,DNA電泳顯示無DNA片段化現象,證實不同鎘濃度所引起的MRC-5細胞凋亡皆為caspase-independent pathway。本實驗室先前的研究結果指出,鎘可藉由產生活性氧分子 (reactive oxygen species)導致細胞凋亡,且粒線體膜電位有去極化 (depolarization)並釋出AIF (apoptosis-inducing factor)的現象。據此,本論文續以共軛焦顯微鏡 (confocal microscope)觀察粒線體內其他凋亡因子的變化,Bax於鎘加入後四小時,由細胞質向粒線體移動,且粒線體膜電位下降,cytochrome c則稍後被釋放出粒線體;鎘處理細胞八小時,AIF (apoptosis-inducing factor)釋出粒線體並集中於細胞核,此為caspase-independent細胞凋亡之重要指標,同時顯示,粒線體為鎘細胞毒性之早期反應胞器。cytochrome c的釋出應可活化caspase 3,並執行細胞凋亡,但直接檢測caspase 3之活性,並未發現其活化,值得再深入探討。鎘經由多種鈣離子通道進入細胞,影響胞內鈣離子含量與訊息傳遞。以Fluo-3 AM染劑觀察細胞內鈣離子的變化,發現鈣離子的濃度在鎘作用後一小時及三小時有五倍的上升。若以細胞內鈣離子螯合劑 (BAPTA-AM)處理細胞,幾乎可完全抑制細胞凋亡。以calpain inhibitor I (ALLN)進行前處理,約可增加50%的細胞存活,但calpain inhibitor II (ALLM),則無保護細胞功能,顯示阻斷鈣離子釋放及其下游-calpain活性可抑制細胞凋亡。以NAC前處理細胞,觀察到一及三小時的鈣離子濃度被有效抑制,顯示ROS為鎘誘導鈣離子增加之主因。綜合上述,本論文說明了,粒線體內的凋亡因子及鈣離子對於鎘所造成的caspase-independent細胞凋亡扮演重要角色。
Studies the Apoptosis Triggered by Cadmium in Human Normal Lung Cells • Environmental pollution by cadmium (Cd) is a worldwide problem due to the increments of industrialization, smoking, and the lack of effective therapy for Cd poisoning. Although the general level of exposure is low, Cd has a long biological half-life of humans, of 10-30 years. It has been reported that Cd causes disorders of renal, skeletal, vascular, and respiratory systems. However, the apoptotic signaling induced by Cd is still not clear. In this study a normal human lung fibroblast (MRC-5) was used as a cell model to investigate the types of cell death induced by Cd using flow cytometry with AnnexinV/PI double staining. The total apoptotic cells reached a plateau of around 40.0% after 24 h exposure of 100 mM Cd. Pretreatment of Z-VAD.fmk, a broad spectrum of caspase inhibitor, could not rescue apoptotic cells from Cd toxicity. Coincidently, we failed to detect 180-bp DNA fragmentation and caspase 3 activation after Cd exposure. These results led to conclude that Cd induced a caspase-independent apoptotic pathway. According to the findings of our previous study, Cd induced 3 folds intracellular ROS burst and the collapse of mitochondria membrane potential, followed by AIF (apoptosis-inducing factor) translocation from the mitochondria to the mucleus. Following this line, the mitochondrial—related apoptotic factors , such as Bax and cytochrome c, were further characterized using confocal microscope. Bax was translocated from the cytoplasm to the mitochondria after 4 h of Cd treatment. The cytochrome c was released from mitochondria at the same time as Bax translocation. After 8 h of Cd treatment, AIF was translocated from the mitochondria into the nucleus. These results demonstrated that mitochondria might be an early target organelle of Cd toxicity. On the other hand, the intracellular calcium ([Ca2+]i) had elevated about 5 folds at 1 and 3 h time points after Cd treatment. The phenomenon was abolished by NAC pretreatment, indication the ROS play a crucial role in Cd-induced elevation of [Ca2+]i. Moreover, the Cd-induced apoptosis was suppressed by pretreatment with intracellular calcium chelator, BAPTA-AM. Calpain inhibitor I (ALLN), but not calpain inhibitor II (ALLM) is able to prevent 50% apoptotic cells from suffering Cd toxicity. This observation supports the notion that calcium-calpain signaling pathway play a pivotal role in Cd-induced apoptosis. In conclusion, combining with our previous results, it is conceivable that Cd induced at least two pathways to conduct caspase-independent apoptosis in MRC-5 cells. One of which is mitochondria-AIF and the other is calcium-calpain pathway. More importantly, we provide several lines of evidence supporting a role for ROS in regulation of caspase-independent cell death triggered by Cd.