Download

1 / 44
440 likes | 608 Views
Class 13 Two sequencing methods that aim to sequence single DNA molecules Pacific Biosciences “zero mode” wave guide Bayley group nanopore method What steps in sequencing methods we considered so far would ability to sequence single molecules avoid?

E N D
Class 13 Two sequencing methods that aim to sequence single DNA molecules Pacific Biosciences “zero mode” wave guide Bayley group nanopore method What steps in sequencing methods we considered so far would ability to sequence single molecules avoid? What technical problem would be eliminated?
Pacific Bioscience seq. strategy – single-molecule, real-time immobilize DNA pol on glass surface at bottom of very small wells (~100nm radius); aim for 1 pol molecule/well add DNA template + primer use dNTPs with fluor attached to terminal P so that it is cleaved off during incorporation (how different from previous fluor-dNTPS we discussed?) collect sequence in real time (enz can go ~1-100b/s)
How long should pulses last? Why would you expect to see only 1 dye at a time?
What steps in previous methods would enzyme- removal of fluor during synthesis eliminate? How fast were previous methods (in terms of bases sequenced/s) given need for chemical steps and washes between base additions?
Challenge for detecting single fluor molecules is usually not sensitivity, but reducing background “Zero mode” waveguide (ZMW)– for well diameters << l w/metallic walls, propagating waves blocked; evanescent waves of exciting and emitted light decay exponentially. Detection depth (volume) ~30nm (10-19 liters) for 100nm holes in aluminum. At mM dye conc., <1 dye/detection vol on average, and any such molecule diffuses out of detection vol in ~100ms (verify: t ~x2/2D, D= 10-12m2/s) whereas dye on dNTP being incorporated by DNA pol expected to be retained by DNA pol. for ms.
Science 299:683, 2003 E-beam lithography makes array of <100nm diam. holes in ~ 100nm thick aluminum film on silica slide For well diameter << l, I(z) ~ e-kz ; excitation of fluor also inhibited by wall proximity; effective illuminated height theoretically ~ 30nm (~ 10x smaller than TIRF) vol~ 10-19 liters
optical set-up to excite and read fluorescence from each well holographic wave plate divides input laser beams into array of beamlets, 1/well ? -> more light/well than If whole field illuminated prism diffracts emitted light to collect diff dye-dNTP signals in different pixels
http://www.sciencemag.org/content/vol0/issue2008/images/data/1162986/DC1/1162986s1.movhttp://www.sciencemag.org/content/vol0/issue2008/images/data/1162986/DC1/1162986s1.mov 93 rows (1mm spacing) x 33 columns (4mm spacing) Light from each well diffracted laterally for diff. detectors
Why do you want high (~mM) dNTP conc.? Binding rate = kon [dNTP] Rate (#/s) at which dNTPs bind each polymerase molecule, per M conc of dNTP; if kon = 107/Ms, what conc. of dNTP do you need for pol to synthesize 10b/s? If you use TIRF, min. vol. of illuminated spot ~pl2*hatt ~p (500nm)2 100nm = 10-19m3 = 10-16liter How many dNTPs in this vol. at 1mM? Need <1 ZMW gets you to <1 by reducing illum. vol~1000-fold!
Idea hinges on using dNTPs with dye labels on phosphate (diff colors for each nt) so dye will be cleaved off during incorporation (don’t need separate chemical step) -> real-time sequencing Base-labeled nucleotide Phosphate-labeled nucleotide
Actual dNTPs used have big dyes + 6 phosphates – They “invented” these dye-NTs and then had to engineer (mutate) DNA pol to use them efficiently
C A T G Emission spectra - would be easiest to distinguish C from G, harder to distinguish 1 from 2, and 3 from 4 (affects “substitution” error rates)
Start with “simple” ss-template – 150 bases with alternating regions with multiple G’s or C’s, synthesize using dGTP-yellow, dCTP-blue, other bases unlabeled
Segment from previous trace Same data in graphical form They at least can pick out the G’s and C’s separated by 0-2 other bases in this region of template, but note variability in peak duration
Now try a circular 75b ss template Their pol enzyme has “strand displacing” activity What advantages might a circular template have? Should you see a pattern repeating every 75 b? What disadvantages would there be to having to use circular DNA templates?
Now try all 4 bases on 150b ss template Note variability in pulse widths
Error rates Single run on 150 b template ~30% of reads incomplete 12 insertions, 8 deletions, 7 mismatches (~20%) What causes apparent insertions? what if base sticks to pol long enough to be detected but falls off before being incorporated, then sticks again and gets incorporated? what could you do about this? – try to engineer enzyme that binds bases tighter (might not work)
What causes apparent deletions? what if base were photobleached before detection? what if enzyme incorporates a base very quickly? If enzyme has constant average rate of incorporation of bound base/unit time, expect Poisson distribution of “hold times”, with largest number of shortest durations
Expected Poisson distribution of hold times They have problem detecting the shortest hold times because of photon counting statistics and noise: dyes produce ~5000 photons/sec -> 50/10ms
What could you do about this problem? try to slow down incorporation rate chemically (they say pH change has a small effect) try to engineer enzyme that incorporates bases more slowly (might not work)
Substitution errors = misreading dyes due to incomplete spectral separation, esp. hard distinguish in short pulses what could you do? – try to improve dyes More immediate solution to high error rates read each DNA template multiple times to generate a consensus sequence (circular templates would be useful…)
They used sequence info from 449 reads of same 150 base template in different wells. Generate a consensus sequence based on random samples of data in which ? each position appears in >15 sequences. Repeat 100x to generate 100 consensus sequences. Error rate in consensus sequences ~2-3%. If they had 3000 wells, why did they only use 449 reads? Suggests they are getting fewer useful wells than they say…
Over-sequencing makes sense if the errors are random but what if the error rate depends on sequence? Summary Many technical hurdles have been overcome, but error rate remains very high Even if they got reliable sequence in real time at rate of 3b/s in each of 3000 wells, would need > 15 days to do human genome 15-fold redundantly, which is competitors’ claimed current rate; 2 yrs ago they said a 1,000,000-well device was coming…
a-hemolysin: heptameric membrane pore-forming protein (bacterial toxin that punches holes in red blood cells) Spontaneously forms 7-mer and inserts into lipid membrane
When inserted in membrane, in electrolyte solution, creates channel that allows ions to cross membrane Can easily detect singlechannels in artificial membrane as they cause step-like changes in current …how many ions go thru pore/sec? 0 pA -20 How much current do you expect from 0.7nm radius pore 10nm long in 1M KCl at 100mV, if conductivity s = 14S/m? I = V/R = VG = VsA/L = .1*14*p*(.7*10-9)2/10-8 = 200pA
How can you make membranes, introduce pores? _ + Add lipid to solution, raise and lower meniscus over hole, 5nm lipid bilayer forms spontaneously (!) 25 µm teflon barrier with ~50mm hole 1 cm Cl- K+ Add a-hemolysin protein to 1 chamber – it inserts itself! 2 nm
b-cyclodextrin: heptameric ring of sugars spontaneously inserts inside a-hemolysin pore stabilized by coordination with 7 identical sites, one in each a-hemolysin monomer
b-CD insertion lowers conductivity W Why might you want to reduce pore diameter? Would you expect charges in pore to influence current? As dNMPs go through pore, they further decrease conductivity; can b-CD be modified so that different bases will -> different decreases in conductivity?
Bayley’s group have made extensive mutations in a-HL and tested many b-CD derivatives to try to make pores that distinguish DNA bases by extent of decr. cond. Here, devise covalent S-S linkage between a-HL and b-CD based on single cys in a-HL and S in modified b-CD in order to have stable small diam. pore How do they get singlecys in heptameric a-HL? Mix 2 types of a-HL, 1 with 1 cys and tail of 8-charged (asp) aa’s; other w/no cys or tail; they form different hetero-7mers; select desired 7mer by electrophoresis
b-CD with -SH group stably associates w/cys-modified a-HL b-CD without S reversibly enters a-HL; with S, it inserts stably but can be removed by reducing -S-S- with DTT
aHL with stably inserted b-CD senses mixture of bases Residual pore currents come in 4 types – why?
More data, with higher conc bases note variable dwell times note minimal overlap in residual pore current histograms – good for distinguishing bases but requires extensive data smoothing
Scatter plot of dwell time vs residual pore current Dwell times have wide distribution, but averages differ for different bases. What does this suggest?
Channel can also distinguish methyl-dCMP, a variant of C associated with silenced gene expression: this system can detect such “epigenetic” changes more easily than other sequencing methods
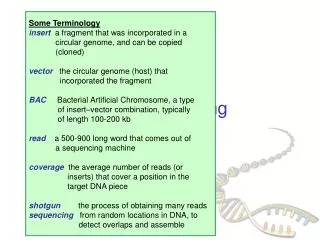
Idea for sequencing – use exonuclease to degrade template to dNMPsand read them going thru pore in the order they are produced
Problem #1: exonuclease doesn’t work in high salt, which is required for good base discrim- ination; they lower salt on side w/exo Test mixed salt system on simpler templates with No A’s shows they can degrade temp- lates with exo’ase and read bases produced No T’s
Technical challenges Reducing [KCL]cis to 200mM allowed exonuclease to work, but decreased ability to distinguish A’s and T’s to ~90%, not good enough for sequencing; they might be able to select exo’s that can work in high salt (e.g. brine bacteria) <tdwell> ~10ms, but Poisson distribution => most dwells are short; for short dwells it is harder to distinguish different bases; very short dwells may -> “deletions” Ability to distinguish 4 bases enhanced by +charge on linker arm; may help to trap bases electrostatically; ? more chemical modifications might improve discrimination
No data yet that exonuclease can be held near enough to pore (e.g. via aHL-exo fusion protein) that that chewed off bases can be read sequentially Will path of some bases (drift + diffusion) be such that they are read out-of-order, or not read at all? Can pore formation be automated and multiplexed? Nevertheless, extraordinary accomplishment in terms of chemical adaptation of nanopore to make real-time, label-free, single-molecule detector that distinguishes 5 bases
Some similarities between solid state FETs and ion channels: charge on channel walls regulates rate charged objects (ions, DNA molecules, peptides) go thru pore if pore is small enough compared to transported object, changes in charge -> exponential changes in transport chips – use simple geometries, Coulomb interactions, engineered structures ~ mm x30nm laterally, a few nm vertically biology – more complex geometries, Coulomb+ chemical interactions (H-bonding, covalent bonds), greater control of nanoscale positioning via protein & DNA engineering but less control at mm scale
Some big ideas from course: Biology at nano-scale is not very different from chemistry, physics at similar scale Biological macromolecules (DNA, protein) allow new forms of engineering and control over nanoscale phenomena Some tools are novel to biology – e.g. replication via pcr Biology provides new things we want to sense (like DNA) and new tools to sense them
At nano-level, single things become detectable Detecting single-molecule events can provide info abt. molecular structure not obtainable in bulk measurements – e.g. whenreplicate molecules cannot be kept in same state for bulk measurements (e.g. phasing problem in DNA sequencing) Aim for detailed and quantitative understanding: how many molecules per sec, unit area, vol; what do they stick to; for how long; how is signal generated; how many photons, etc Think critically as you learn!
Suggestions for student presentations Go over paper with me beforehand if at all possible! Stick to a few main points – you have only ~20 minutes Try to teach us something interesting you learned