Download
1 / 1
20 likes | 174 Views
Materials Computation Center, University of Illinois Duane Johnson and Richard Martin, NSF DMR-03-25939 Accurate Quantum Chemistry via Machine-Learning & Evolutionary Algorithms Duane D. Johnson (MatSE), David E. Goldberg (GE), Todd J. Martinez (Chem),

E N D
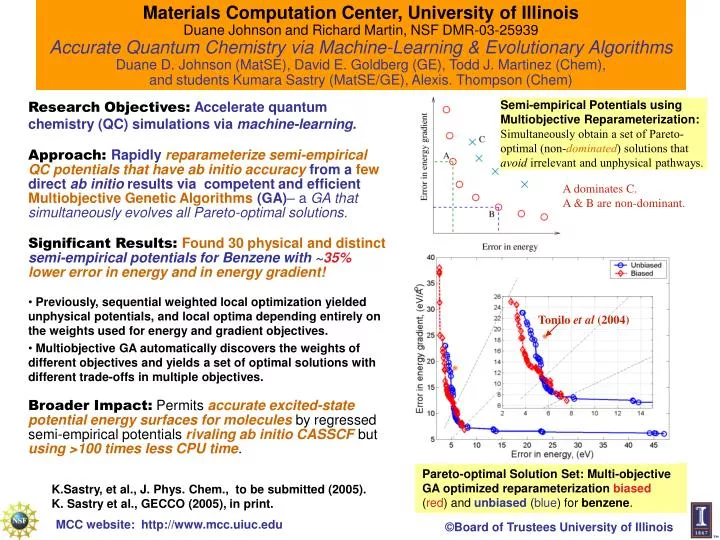
Materials Computation Center, University of Illinois Duane Johnson and Richard Martin, NSF DMR-03-25939Accurate Quantum Chemistry via Machine-Learning & Evolutionary Algorithms Duane D. Johnson (MatSE), David E. Goldberg (GE), Todd J. Martinez (Chem), and students Kumara Sastry (MatSE/GE), Alexis. Thompson (Chem) Tonilo et al (2004) • ResearchObjectives:Accelerate quantum chemistry (QC) simulationsvia machine-learning. • Approach: Rapidly reparameterize semi-empirical QC potentialsthat have ab initio accuracy from a few direct ab initio resultsvia competent and efficient Multiobjective Genetic Algorithms (GA)– a GA that simultaneously evolves all Pareto-optimal solutions. • Significant Results: Found 30 physical and distinct semi-empirical potentials for Benzene with ~35%lower error in energy and in energy gradient! • Previously, sequential weighted local optimization yielded unphysical potentials, and local optima depending entirely on the weights used for energy and gradient objectives. • Multiobjective GA automatically discovers the weights of different objectives and yields a set of optimal solutions with different trade-offs in multiple objectives. • Broader Impact: Permits accurate excited-state potential energy surfaces for molecules by regressed semi-empirical potentials rivaling ab initio CASSCF but using >100 times less CPU time. Semi-empirical Potentials using Multiobjective Reparameterization:Simultaneously obtain a set of Pareto-optimal (non-dominated) solutions that avoid irrelevant and unphysical pathways. • A dominates C. • A & B are non-dominant. Pareto-optimal Solution Set: Multi-objective GA optimized reparameterization biased (red) and unbiased (blue) for benzene. K.Sastry, et al., J. Phys. Chem., to be submitted (2005). K. Sastry et al., GECCO (2005), in print.