Download
1 / 1
10 likes | 132 Views
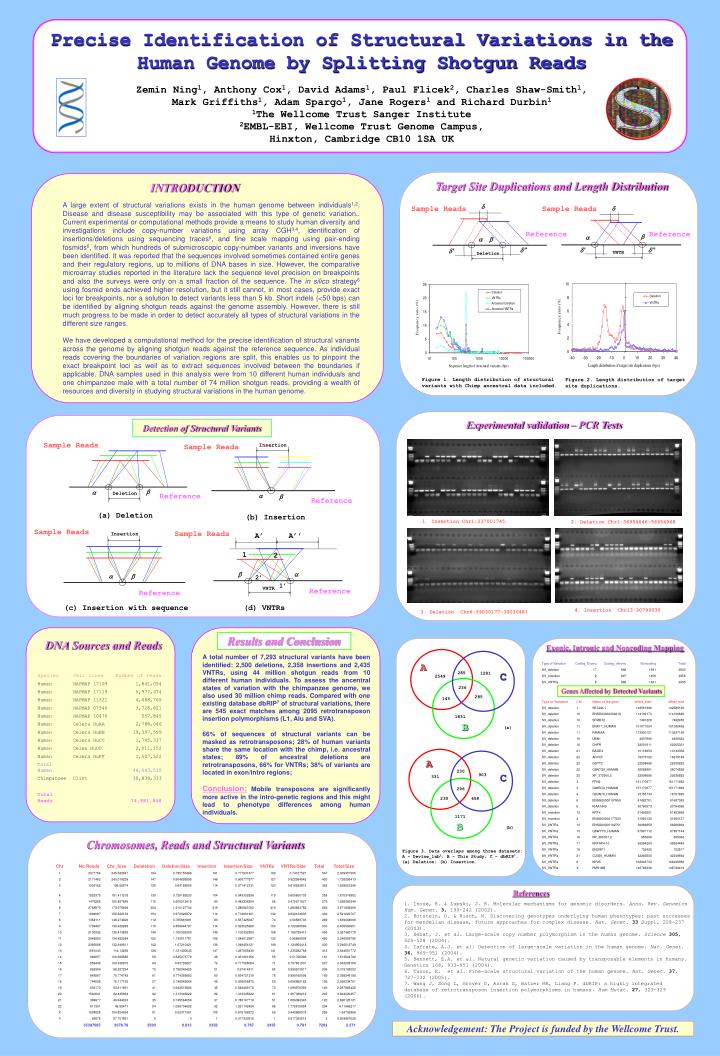
S. Detection of Structural Variants. Sample Reads. Insertion. Sample Reads. b. a. Deletion. Reference. a. b. Reference. (a) Deletion. (b) Insertion. Sample Reads. Sample Reads. Insertion. A’. A’’. d. Sample Reads. Sample Reads. d. 1. 2. b. a. a. b. 2’. Reference.

E N D
S Detection of Structural Variants Sample Reads Insertion Sample Reads b a Deletion Reference a b Reference (a) Deletion (b) Insertion Sample Reads Sample Reads Insertion A’ A’’ d Sample Reads Sample Reads d 1 2 b a a b 2’ Reference Reference b a a b 1’ VNTR Reference Reference d’’ d’ d’’ d’ VNTR Deletion (c) Insertion with sequence (d) VNTRs A A 230 285 1281 C 903 C 2549 331 236 296 285 145 230 658 1831 1171 B B (b) Precise Identification of Structural Variations in the Human Genome by Splitting Shotgun Reads Zemin Ning1, Anthony Cox1, David Adams1, Paul Flicek2, Charles Shaw-Smith1, Mark Griffiths1, Adam Spargo1, Jane Rogers1 and Richard Durbin1 1The Wellcome Trust Sanger Institute 2EMBL-EBI, Wellcome Trust Genome Campus, Hinxton, Cambridge CB10 1SA UK Target Site Duplications and Length Distribution INTRODUCTION A large extent of structural variations exists in the human genome between individuals1,2. Disease and disease susceptibility may be associated with this type of genetic variation.. Current experimental or computational methods provide a means to study human diversity and investigations include copy-number variations using array CGH3,4, identification of insertions/deletions using sequencing traces5, and fine scale mapping using pair-ending fosmids6, from which hundreds of submicroscopic copy-number variants and inversions have been identified. It was reported that the sequences involved sometimes contained entire genes and their regulatory regions, up to millions of DNA bases in size. However, the comparative microarray studies reported in the literature lack the sequence level precision on breakpoints and also the surveys were only on a small fraction of the sequence. The in silico strategy6 using fosmid ends achieved higher resolution, but it still cannot, in most cases, provide exact loci for breakpoints, nor a solution to detect variants less than 5 kb. Short indels (<50 bps) can be identified by aligning shotgun reads against the genome assembly. However, there is still much progress to be made in order to detect accurately all types of structural variations in the different size ranges. We have developed a computational method for the precise identification of structural variants across the genome by aligning shotgun reads against the reference sequence. As individual reads covering the boundaries of variation regions are split, this enables us to pinpoint the exact breakpoint loci as well as to extract sequences involved between the boundaries if applicable. DNA samples used in this analysis were from 10 different human individuals and one chimpanzee male with a total number of 74 million shotgun reads, providing a wealth of resources and diversity in studying structural variations in the human genome. Figure 1. Length distribution of structural variants with Chimp ancestral data included. Figure 2. Length distribution of target site duplications. Experimental validation – PCR Tests 1. Insertion Chr1:237001745 2. Deletion Chr1:56954646-56954968 4. Insertion Chr13:30790030 3. Deletion Chr6:39030177-39030481 Results and Conclusion DNA Sources and Reads Exonic, Intronic and Noncoding Mapping A total number of 7,293 structural variants have been identified: 2,500 deletions, 2,358 insertions and 2,435 VNTRs, using 44 million shotgun reads from 10 different human individuals. To assess the ancentral states of variation with the chimpanzee genome, we also used 30 million chimp reads. Compared with one existing database dbRIP7 of structural variations, there are 545 exact matches among 2095 retrotransposon insertion polymorphisms (L1, Alu and SVA). 66% of sequences of structural variants can be masked as retrotransposons; 28% of human variants share the same location with the chimp, i.e. ancestral states; 89% of ancestral deletions are retrotransposons, 66% for VNTRs; 38% of variants are located in exon/intro regions; Conclusion: Mobile transposons are significantly more active in the intro-genetic regions and this might lead to phenotype differences among human individuals. Genes Affected by Detected Variants (a) Chromosomes, Reads and Structural Variants Figure 3. Data overlaps among three datasets: A – Devine_lab5; B – This Study; C – dbRIP7. (a) Deletion; (b) Insertion. References 1. Inoue, K. & Lupski, J. R. Molecular mechanisms for genomic disorders. Annu. Rev. Genomics Hum. Genet.3, 199–242 (2002). 2. Botstein, D. & Risch, N. Discovering genotypes underlying human phenotypes: past successes for mendelian disease, future approaches for complex disease. Nat. Genet.33 Suppl, 228–237 (2003). 3. Sebat, J. et al. Large-scale copy number polymorphism in the human genome. Science305, 525–528 (2004). 4. Iafrate, A.J. et al. Detection of large-scale variation in the human genome. Nat. Genet. 36, 949–951 (2004). 5. Bennett, E.A. et al. Natural genetic variation caused by transposable elements in humans. Genetics 168, 933-951 (2004). 6. Tuzun, E. et al. Fine-scale structural variation of the human genome. Nat. Genet.37, 727–732 (2005). 7. Wang J, Song L, Grover D, Azrak S, Batzer MA, Liang P. dbRIP: a highly integrated database of retrotransposon insertion polymorphisms in humans. Hum Mutat. 27, 323-329 (2006). Acknowledgement: The Project is funded by the Wellcome Trust.






