Download
1 / 1
20 likes | 94 Views
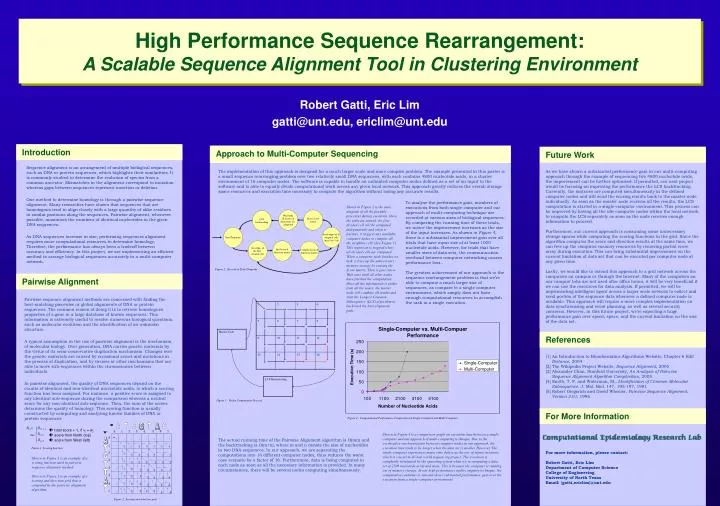
A. T. C. G. T. A. C. w. 1. 2. 3. 4. 5. 6. 7. 0. 0. v. A. 1. T. 2. 0. G. 3. 0. T. 4. 0. T. 5. 0. S i,j = S i-1, j-1 max S i-1, j S i, j-1. A. 6. 0. {. T. 7. 0. 0. High Performance Sequence Rearrangement:

E N D
A T C G T A C w 1 2 3 4 5 6 7 0 0 v A 1 T 2 0 G 3 0 T 4 0 T 5 0 Si,j = Si-1, j-1 max Si-1, j Si, j-1 A 6 0 { T 7 0 0 High Performance Sequence Rearrangement: A Scalable Sequence Alignment Tool in Clustering Environment Modelling Tuberculosis: A Scalable Agent-Based Framework Robert Gatti, Eric Lim gatti@unt.edu, ericlim@unt.edu Introduction Approach to Multi-Computer Sequencing Future Work Sequence alignment is an arrangement of multiple biological sequences, such as DNA or protein sequences, which highlights their similarities. It is commonly studied to determine the evolution of species from a common ancestor. Mismatches in the alignment correspond to mutation whereas gaps between sequences represent insertion or deletion. One method to determine homology is through a pairwise sequence alignment. Many researches have shown that sequences that are homologous tend to align closely with a large quantity of alike residues in similar positions along the sequences. Pairwise alignment, whenever possible, maximizes the numbers of identical nucleotides in the given DNA sequences. As DNA sequences increase in size, performing sequences alignment requires more computational resources to determine homology. Therefore, the performance has always been a tradeoff between accuracy and efficiency. In this project, we are implementing an efficient method to arrange biological sequences accurately in a multi-computer network. The implementation of this approach is designed for a much larger scale and more complex problem. The example presented in this poster is a small sequence rearranging problem over two relatively small DNA sequences, with each contains 4800 nucleotide acids, in a cluster environment of 16 computer nodes. The software is capable to handle an unlimited computer nodes defined as a set of an input to the software and is able to equally divide computational work across any given local network. This approach greatly reduces the overall storage space resources and execution time necessary to compute the algorithm without losing any accurate results. As we have shown a substantial performance gain in our multi-computing approach through the example of sequencing two 4800 nucleotide acids, the improvement can be further optimised. If permitted, our next project would be focusing on improving the performance the LCS backtracking. Currently, the matrices are computed simultaneously in the defined computer nodes and will send the scoring results back to the master node individually. As soon as the master node receives all the results, the LCS computation is started in a single-computer environment. This process can be improved by having all the idle computer nodes within the local network to compute the LCS separately as soon as the node receives enough information to proceed. Furthermore, our current approach is consuming some unnecessary storage spaces while computing the scoring functions in the grid. Since the algorithm computes the score and direction results at the same time, we can free up the computer memory resources by removing partial score array during execution. This can bring substantial improvement on the current limitation of data set that can be executed per computer node at any given time. Lastly, we would like to extend this approach to a grid network across the computers on campus or through the Internet. Many of the computers on our campus’ labs are not used after office hours, it will be very beneficial if we can use the resources for data analysis. If permitted, we will be implementing intelligent agent across a larger scale network to collect and send portion of the sequence data whenever a defined computer node is available. This approach will require a more complex implementation on data synchronizing and event planning, as well as several security concerns. However, in this future project, we’re expecting a huge performance gain over speed, space, and the current limitation on the size of the data set. To analyse the performance gain, numbers of executions from both single-computer and our approach of multi-computing technique are recorded at various sizes of biological sequences. By comparing the running time of these trials, we notice the improvement increases as the size of the input increases. As shown in Figure 4, there is a substantial improvement gain over all trials that have input size of at least 1000 nucleotide acids. However, for trials that have smaller sizes of data sets, the communication overhead between computer networking causes performance loss.. The greatest achievement of our approach to the sequence rearrangement problem is that we’re able to compute a much larger size of sequences, as compare to a single computer environment, which simply does not have enough computational resources to accomplish the task in a single execution. Shown in Figure 2 is the state diagram of all the possible processes during execution. Once the software started, the first divided cell will be computed independently and when it finishes, it triggers any available computer nodes to compute all the neighbor cells (See Figure 3). This repetition is stopped when all divided cells are computed. When a computer node finishes its task, it frees up the unnecessary memory storage by erasing the Score matrix. Then it goes into a Wait state until all other nodes have finished the computation. Once all the information is gather from all the nodes, the master node will combine all results and start the Longest-Common-Subsequence (LCS) algorithm to backtrack the best alignment path. Wait until all Scores & Directions are computed Erase Score matrix LCS backtracking Start/Terminate Send edges of all diagonal cells (m,n-1)(n-1,m) Init Score & Direction matrix Get edges of the first divided cell Compute Score & Direction matrix Figure 2 : Execution State Diagram Pairwise Alignment Pairwise sequence alignment methods are concerned with finding the best-matching piecewise or global alignments of DNA or protein sequences. The common reason of doing it is to retrieve homologues properties of a gene in a large database of known sequences. This information is extremely useful to resolve numerous biological questions, such as molecular evolution and the identification of an unknown structure. A typical assumption in the use of pairwise alignment is the mechanism of molecular biology. Over generation, DNA carries genetic materials by the virtue of its semi-conservative duplication mechanism. Changes over the genetic materials are caused by occasional errors and mutations in the process of duplication, and by viruses or other mechanisms that are able to move sub-sequences within the chromosomes between individuals. In pairwise alignment, the quality of DNA sequences depend on the counts of identical and non-identical nucleotide acids, to which a scoring function has been assigned. For instance, a positive score is assigned to any identical sub-sequence during the comparison whereas a neutral score for any non-identical sub-sequence. Then, the sum of the scores determine the quality of homology. This scoring function is usually constructed by computing and analysing known families of DNA or protein sequences. References [1] An Introduction to Bioinformatics Algorithms Website, Chapter 6 Edit Distance, 2004 [2] The Wikipedia Project Website, Sequence Alignment, 2005 [3] Alexander Chan, Stanford University, An Analysis of Pairwise Sequence Alignment Algorithm Complexities, 2005 [4] Smith, T. F. and Waterman, M., Identification of Common Molecular Subsequence. J. Mol. Biol. 147, 195-197, 1981 [5] Robert Giegerich and David Wheeler, Pairwise Sequence Alignment, Version 2.01, 1996 Figure 3 : Nodes Computation Process For More Information Figure 4 : Computational Performance Comparison on Single-Computer and Multi-Computer total score + 1, if vi = wj score from North (top) score from West (left) Shown in Figure 4 is a comparison graph on execution time between a single-computer and our approach of multi-computing technique. Due to the overhead in synchronization between computer nodes in our approach, the execution time tends to be longer when the data set is smaller. However, The single-computer experiences major time delays as the size of inputs increases, which is crucial in all real world sequencing project. The execution is completely terminated by the operating system when we’re computing a data set of 2500 nucleotide acids and more. This is because the computer is running out of memory storage. In our high performance multi-computer technique, the computation continues to run and shows substantial performance gain over the execution from a single-computer environment. The actual running time of the Pairwise Alignment algorithm is O(mn) and the backtracking is O(m+n), where m and n denote the size of nucleotides in two DNA sequences. In our approach, we are separating the computations into 16 different computer nodes, thus reduces the worst case scenario by a factor of 16. Furthermore, data is being computed in each node as soon as all the necessary information is provided. In many circumstances, there will be several nodes computing simultaneously. For more information, please contact: Robert Gatti, Eric Lim Department of Computer Science College of Engineering University of North Texas Email: {gatti,ericlim}@unt.edu 0 0 0 0 0 0 0 0 1 1 1 1 1 1 1 Figure 1: Scoring function 1 Shown in Figure 1 is an example of a scoring function used in pairwise sequence alignment method. Shown in Figure 2 is an example of a scoring and direction grid that is computed by the pairwise alignment algorithm. 2 2 2 2 2 2 1 2 2 3 3 3 3 1 2 2 3 4 4 4 1 2 2 3 4 4 4 1 2 2 3 4 5 5 1 2 2 3 4 5 5 Figure 2: Scoring and direction grid