Download

1 / 21

E N D
COAGULATION Dr. Hasan Fahmawi, MRCP(UK), FRCP(Edin)
Haemostasis-blood must be maintained in a fluid state in order to function as a transport system, but must be able to solidify to form a clot following vascular injury in order to prevent excessive bleeding, a process known as haemostasis. It is localised to the tissue damage and is followed by removal of the clot and tissue damage. • It is achieved by complex interaction between the vascular endothelium, platelets, von Willebrand factor, coagulation factor, natural anticoagulant, and fibrinolytic enzymes. Dysfunction of any of these components may result in haemorrhage or thrombosis Platelets- formed in the bone marrow from megakaryocytes, which are stimulated by thrombopoietin produced in the liver. They circulate in the blood for 8-10 days. Some 30% are pooled in the spleen and don’t circulate, drugs that inhibit platelets-Aspirin inhibit cyclo-oxygenase, Clopidogrel-ADP, dipyridamole-phosphodiesterase inhibitor, and Glycoprotein 11b/11a inhibitors abciximab, which prevent fibrinogen binding.
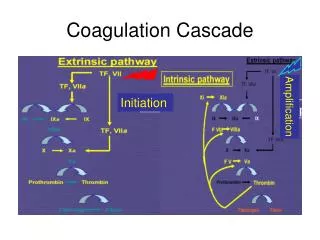
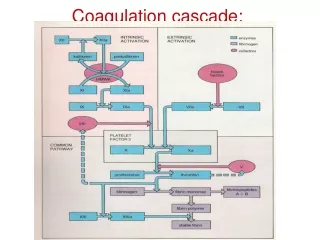
Coagulation factors • Consist of a cascade of inactive zymogen proteins designated by Roman numerals. Activated factors are designated by the suffix a, • Some of these reactions require phospholipid and Ca , Coagulation initiated by extrinsic or tissue factor and amplified by the intrinsic pathway. • Coagulation factors are synthesized by the liver, although factor V is also produced by platelets and endothelial cells. Vit. k dependent factor are -1 9 7 2 • Causes of coagulation factors deficiency are either congenital e.g.(haemophilia) and acquired e.g.(liver failure), which causes bleeding. Congenital factors are due to decrease synthesis, vWD disease is the most common inherited deficiency, acquired may be due to under-production(liver disease), increased consumption(DIC), inhibition of function(heparin)or immune inhibitors of coagulation(acquired haemophilia A).
Haemophilia A • The factor V111 gene is located on the X chromosome, all daughters are obligate carriers, antenatal diagnosis by chorionic villous sampling is possible in families of known mutations. • Haemophilia breeds true within a family, all members have the same factor V111 gene mutation and similarly severe or mild phenotype. • Female carrier may have reduced factor V111 level because of random inactivation of their X chromosome. This can result in a mild bleeding disorder, thus all known or suspected carriers should have their factor V111 level measured.
+Clinical features • Depend on factor V111 level, in severe forms spontaneous bleeding into skin, muscles and joints, as well as retroperitoneal and intracranial bleeding. • The major morbidity of recurrent bleeding in severe haemophilia is musculoskeletal. Large joints especially, elbows, knees, ankles, and hips are involved. Muscle haematomas are also characteristic, mostly in the calf and psoas muscles, if early treatment is not given a hot , swollen, and very painful joint or muscle haematoma develops. Recurrent bleeding into joints leads to synovial hypertrophy, destruction of cartilage and chronic haemophilic arthropathy. A large psoas bleed may extend to compress femoral nerve, calf haematoma may increase pressure within the inflexible fascial sheath, causing compartment syndrome with ischaemic necrosis, fibrosis and subsequent contraction and shortening of the Achilles tendon.
Treatment • Prophylaxis-aim to maintain trough levels 0.02U/ml, so reducing bleeding episodes for men with severe haemophilia. This reduces the deterioration of joints which is the major long term morbidity. • Half life is 12 hrs. Recombinant factor concentrates are virus free, and long acting, but costly. • In less affluent countries factor V111 concentrate is used to treat bleeding, screened for hepatitis A, B, C, and HIV. • Bed rest or splint reduces continuing haemorrhage, as it stops mobility and physiotherapy is started to strengthen surrounding muscles. All patient should be offered hepatitis A and B vaccination. • Vasopressin receptor agonist desmopressin is used in moderate and mild cases.(monitor water retention which can result in significant hyponatraemia). vCJD might be transmitted. Anti-factor V111 antibodies which arise in 20% of those with severe haemophilia, such antibodies rapidly neutralize therapeutic infusions making them ineffective. Infusion of activated clotting factors e.g. factor V11a or factor V11 inhibitor bypassing activity (FEIBA)may stop bleeding.
Haemophilia B • X- linked inheritance of factor 1X deficiency, clinically indistinguishable from haemophilia A but is less common. • PEGylated forms of factor 1X are available, which are given every one or even two wks. Less than 1% develop antibodies, when this does occur, however, it may be heralded by a severe allergic reaction.
Von Willebrand disease • Common but usually mild bleeding disorder, this protein is synthesised by endothelial cells and megakaryocytes, and is involved in both platelets function and coagulation. It acts as carrier for factor V111, it’s deficiency lowers factor V111. vWF forms bridges between platelets and collagen allowing platelets to adhere to vessel wall. Blood group antigens (A and B) are expressed on vWF reducing it’s susceptibility for proteolysis, people with blood group O has lower levels.
Clinical features • Haemorrhagic manifestations similar to platelet dysfunction. Superficial bruising, epistaxis, menorrhagia and GI bleeding are common. Bleeding episodes are much less frequent than in severe haemophilia, and excessive haemorrhage may be observed only after trauma or surgery. Within single family the disease has variable penetrance some bleed severely, others are a symptomatic. • Management-minor bleeds desmopressin, and in mucosal bleeding tranexamic acid. In persistent bleeding factor V111 concentrate is used. Bleeding in type B patients respond to factor V111/vWF concentrate. • Factor X111 deficiency is associated with recurrent fetal death. • Factor X1 deficiency-severe bleeding confined to patients with level below 15%. • Deficiency of factor V11, X and X111 is associated with severe bleeding. • Factor X11 is asymptomatic but cause prolonged clotting time in the test tube.
Acquired bleeding disorder • Liver disease-associated with bleeding and venous thrombosis, due to impaired synthesis of procoagulant factors, reduced production of natural anticoagulants and reduced antifibrinolytic activities. • Reduced factors V, V11, 1X, X, X1, prothrombin and fibrinogen. Clearance of fibrinogen activator is reduced. In obstructive jaundice there is reduced vit. K absorption . Thrombocytopaenia due to hypersplenism. • Renal failure-The severity of bleeding is proportional to the plasma urea concentration. Bleeding manifestations are those of platelets dysfunction, GI in particular. Causes are multifactorial, anaemia, thrombocytopaenia and the accumulation of low molecular weight waste products.
Thrombophilia • Antithrombin deficiency, autosomal dominant, around 70% will develop VTE before the age of 60 years. Pregnancy is a high risk and requires high dose LMW heparin prophylaxis. Antithrombin concentrate is available. • Protein C and S deficiency, protein C and its co-factor protein S are vit. K dependent natural anticoagulants. Five fold relative risk of VTE. • Factor V, Leiden, risk of thrombosis is five in heterozygous and 50 in rare homozygous, present in 5% of Europeans. • Prothrombin G20210A, result in increased plasma level of prothrombin leading to 2-3 fold increase in risk of VTE. .
Antiphospholipid syndrome • Anticardiolipin antibody - ELIZA test, these usually contain/ Beta 2 glycoprotein 1. • Lupus anticoagulant, increased APTT. Patients who are triple positive have an increased risk o thrombotic events • Recurrent abortions, VTE and arterial thrombosis(stroke) which requires warfarin for treatment. VTE requires long term treatment after first event. In pregnancy aspirin and heparin are given.
DIC • It is characterised by systemic activation of the pathways involved in coagulation and its regulation, this may result in intravascular generation of fibrin clots causing multi-organ failure, with simultaneous coagulation and platelet consumption, causing bleeding. • Investigation-PT, APTT, Fibrinogen, platelet count and FDPs, • Associated with acidosis, dehydration and renal failure, which must be treated, besides underlying cause. In bleeding FFP and platelets are given. Thrombosis treated with unfractionated heparin unless clearly contraindicated.