Download
1 / 1
10 likes | 155 Views
Characterizing Constraints Acting On HIV-1 Viral Evolution. Jeongmin Woo, Simon C. Lovell and David L. Robertson

E N D
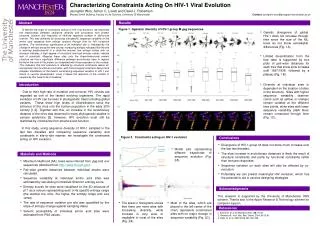
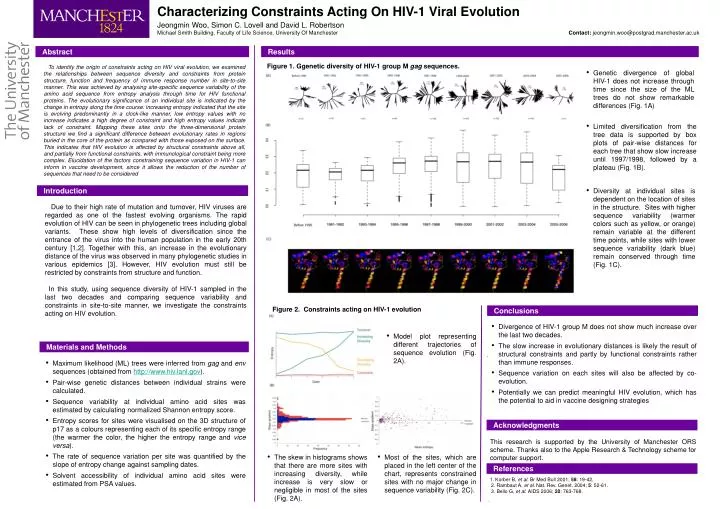
Characterizing Constraints Acting On HIV-1 Viral Evolution Jeongmin Woo, Simon C. Lovell and David L. Robertson Michael Smith Building, Faculty of Life Science, University Of Manchester Contact: jeongmin.woo@postgrad.manchester.ac.uk Abstract Results Figure 1. Ggenetic diversity of HIV-1 group M gag sequences. To identify the origin of constraints acting on HIV viral evolution, we examined the relationships between sequence diversity and constraints from protein structure, function and frequency of immune response number in site-to-site manner. This was achieved by analysing site-specific sequence variability of the amino acid sequence from entropy analysis through time for HIV functional proteins. The evolutionary significance of an individual site is indicated by the change in entropy along the time course: increasing entropy indicated that the site is evolving predominantly in a clock-like manner, low entropy values with no increase indicates a high degree of constraint and high entropy values indicate lack of constraint. Mapping these sites onto the three-dimensional protein structure we find a significant difference between evolutionary rates in regions buried in the core of the protein as compared with those exposed on the surface. This indicates that HIV evolution is affected by structural constraints above all, and partially from functional constraints, with immunological constraint being more complex. Elucidation of the factors constraining sequence variation in HIV-1 can inform in vaccine development, since it allows the reduction of the number of sequences that need to be considered • Genetic divergence of global HIV-1 does not increase through time since the size of the ML trees do not show remarkable differences (Fig. 1A) • Limited diversification from the tree data is supported by box plots of pair-wise distances for each tree that show slow increase until 1997/1998, followed by a plateau (Fig. 1B). Introduction • Diversity at individual sites is dependent on the location of sites in the structure. Sites with higher sequence variability (warmer colors such as yellow, or orange) remain variable at the different time points, while sites with lower sequence variability (dark blue) remain conserved through time (Fig. 1C). Due to their high rate of mutation and turnover, HIV viruses are regarded as one of the fastest evolving organisms. The rapid evolution of HIV can be seen in phylogenetic trees including global variants. These show high levels of diversification since the entrance of the virus into the human population in the early 20th century [1,2]. Together with this, an increase in the evolutionary distance of the virus was observed in many phylogenetic studies in various epidemics [3]. However, HIV evolution must still be restricted by constraints from structure and function. In this study, using sequence diversity of HIV-1 sampled in the last two decades and comparing sequence variability and constraints in site-to-site manner, we investigate the constraints acting on HIV evolution. Figure 2. Constraints acting on HIV-1 evolution Conclusions • Divergence of HIV-1 group M does not show much increase over the last two decades. • The slow increase in evolutionary distances is likely the result of structural constraints and partly by functional constraints rather than immune responses. • Sequence variation on each sites will also be affected by co-evolution. • Potentially we can predict meaningful HIV evolution, which has the potential to aid in vaccine designing strategies • Model plot representing different trajectories of sequence evolution (Fig. 2A). . Materials and Methods • Maximum likelihood (ML) trees were inferred from gag and env sequences (obtained from http://www.hiv.lanl.gov). • Pair-wise genetic distances between individual strains were calculated. • Sequence variability at individual amino acid sites was estimated by calculating normalized Shannon entropy score. • Entropy scores for sites were visualised on the 3D structure of p17 as a colours representing each of its specific entropy range (the warmer the color, the higher the entropy range and vice versa). • The rate of sequence variation per site was quantified by the slope of entropy change against sampling dates. • Solvent accessibility of individual amino acid sites were estimated from PSA values. Acknowledgments This research is supported by the University of Manchester ORS scheme. Thanks also to the Apple Research & Technology scheme for computer support. • Most of the sites, which are placed in the left center of the chart, represents constrained sites with no major change in sequence variability (Fig. 2C). • The skew in histograms shows that there are more sites with increasing diversity, while increase is very slow or negligible in most of the sites (Fig. 2A). References 1. Korber B, et al. Br Med Bull 2001; 58: 19-42. 2. Rambaut A, et al. Nat. Rev. Genet. 2004; 5: 52-61. 3. Bello G, et al. AIDS 2006; 20: 763-768. .