Download
1 / 45
460 likes | 781 Views
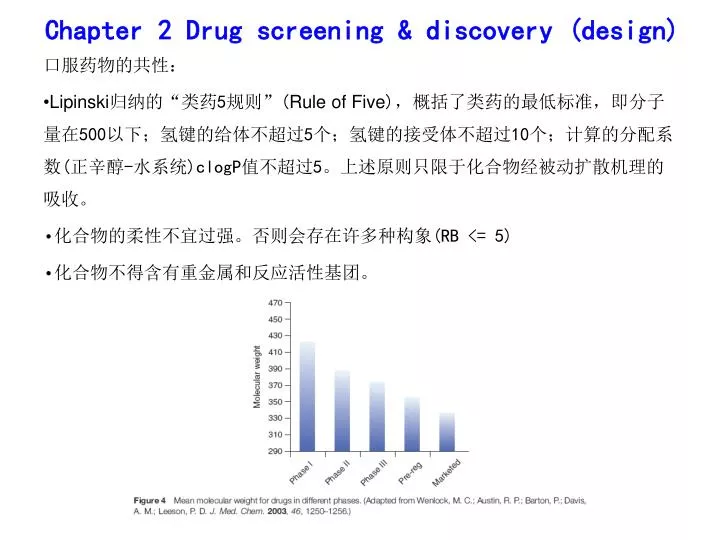
Chapter 2 Drug screening & discovery (design). 口服药物的共性: Lipinski 归纳的 “ 类药 5 规则 ” ( Rule of Five ) ,概括了类药的最低标准,即分子量在 500 以下;氢键的给体不超过 5 个;氢键的接受体不超过 10 个;计算的分配系数 ( 正辛醇 - 水系统 )clogP 值不超过 5 。上述原则只限于化合物经被动扩散机理的吸收。 化合物的柔性不宜过强。否则会存在许多种构象 (RB <= 5) 化合物不得含有重金属和反应活性基团。. 药代动力学性质.

E N D
Chapter 2 Drug screening & discovery (design) • 口服药物的共性: • Lipinski归纳的“类药5规则”(Rule of Five),概括了类药的最低标准,即分子量在500以下;氢键的给体不超过5个;氢键的接受体不超过10个;计算的分配系数(正辛醇-水系统)clogP值不超过5。上述原则只限于化合物经被动扩散机理的吸收。 • 化合物的柔性不宜过强。否则会存在许多种构象(RB <= 5) • 化合物不得含有重金属和反应活性基团。
药代动力学性质 • 临床试验被终止淘汰的候选药物40%(1990s)是由于药代动力学不合理造成的 • 决定药物能够穿越细胞膜并在胞浆中转运的性质是分子的化学结构,表现在分子量,离解常数,亲脂性,极性表面积,以及形成氢键的数目等 • 药物的代谢转化主要在肝脏中发生。将细胞色素P450 2D6和3A4催化中心的三维结构作为药效团,可用于预测未知化合物的代谢命运。通过分析化合物的三维结构与半衰期的相关性,可以来预测未知物的代谢模式 (近20年药代药动学进步显著!) 毒性的预测 • 基于已有化合物的毒性和结构特征,经线性判别分析和多重回归分析得到的模型,可用来预测未知物的毒性。 • 基于知识的专家系统(knowledge-based system)的软件如DEREK,可批处理化合物的致癌性、致畸性、致突变性、刺激性、皮肤敏感性、急性毒性和神经毒等。另一个基于知识的专家系统是HazardExpert程序,通过输入化合物名称、给药途径、剂量和用药时程,程序可给出结果。
基于结构的设计 • 在受体结构信息已知的情况下,可根据结合部位的三维结构信息,用分子对接方法,对互补性好、评分高的化合物,可预计有较强的亲和力。若不知受体的三维结构,可根据药效团特征筛选虚拟库,并以不同程度的限制条件,“滤除”与药效团无相似性的分子。 知识产权的预测 • 化合物具备自主的知识产权和专利保护(IP)的前景,是开发决策的重要指标,筛选虚拟库和组合库时要剔除已被其它专利覆盖或有可能侵权的化合物。所以,完备的化合物检索查新系统可确保化合物结构的新颖性。 (emolecules.com)
先导化合物发现 • 天然活性物质 • 高通量筛选 • 基于配体或底物的分子设计 • 基于药物的副作用 • 基于代谢作用 • 幸运Serendipity • 基于片段的药物发现
1.天然生物活性物质作为先导物 1、天然活性物质直接作药物:利血平,万古霉素(糖肽类抗生素) Reserpine:肾上腺素能神经元阻断性 抗高血压药,提取于萝芙木属多种植物 2、天然活性物质作先导物,适当分子变换:紫杉醇,长春碱 Paclitaxel: 促进微管蛋白聚合,阻止其在有丝分裂的功能,治疗卵巢癌、乳腺癌、恶性黑色素瘤。 docetaxel:水溶性增加,活性〉紫杉醇,无交叉耐药
3、改造天然产物 Km=10mM LDL-C: 引起动脉粥状样硬化、心肌梗死 桔青霉Penicillium citrinum提纯得到Lovastatin Ki = 1nM Lovastatin
statins Fluvastatin Pitavastatin(Nisvastatin) Atorvastatin (Lipitor, best sale)
青蒿素 黄花蒿 Artemisia annula 生物利用度较低 复发率高 青蒿素 Artemisinin 蒿甲醚 Artemether (Novartis PCT)
喜树(中国) Camptotheca acuminata • 喜树碱 水溶性较差,毒性大 羟基喜树碱 Hydroxycamptothecin 拓扑替康 Topotecan (GSK 2007) 作用于DNA topoisomerase I (化疗药物) 治疗SCLC,卵巢癌,结肠癌
南美洲古柯 Erythroxylum coca Lam • 局麻药 可卡因 Cocaine 普鲁卡因 Procaine (1905) Lidocaine (1943 Synthesis; 1949 on market)
动物毒素 • 蛇毒Bungarotoxin,N2受体拮抗剂肌松药 • 蛇毒Batroxobin,溶血栓酶抗栓药 • 鱼毒Tetrodotoxin,钠通道阻断剂心血管药物 • 蜂毒Apamin,钙通道阻断剂和钾通道开放剂心血管药物
2、高通量筛选(HTS) Cons: Low hit rate: 0.001% Low druggability High false positive rate 多靶标对化合物库的筛选(目前的主要筛选方法) 立体筛选方法的灵敏性、微量化、自动化 大容量多样性化合物库 组合化学Combinatorial chemistry • 同时制备含众多分子的化合物库 • 以代数级数增加构建块的数目,库容量则以几何级数增加 • 与高通量筛选(high-throughput screening, HTS)技术结合,可极大地加快先导物发现和优化的速度 • 平行合成和混分合成 • 固相合成和液相合成 • 小分子组合合成 • 计算机辅助设计及虚拟库合成
组合生物合成Combinatorial biosynthesis • 基本原理 基因变异(混合、匹配、交换、突变等) 基因克隆 多种变异的酶系 多种非天然的天然物质
组合生物催化Combinatorial biocatalysis • 基本原理 变异酶系或微生物酶系 催化小分子化合物转化 多种人工的天然化合物
3、基于配体或底物的分子设计 • 酶反应过程:酶抑制剂 • 酶结构 • 底物、过渡态、产物结构 • ACEI、COX-2、GABA-T、MAO抑制剂等 • 抗代谢物:酶抑制剂,致死合成 • 与受体作用过程:激动剂或拮抗剂 • 受体结构 • 配体结构 • 肾上腺素能药物、胆碱能药物、甾体药物等
5-hydroxytryptamine (5-HT):神经递质,低水平--〉偏头痛 5-HT receptor isomers: 5-HT1, 5-HT2, 5-HT3 Activity(5-HT1) = 2 (vs. 5-HT) Activity(5-HT2) = 1/25 Sumatriptan (GSK,2009 expired):Activity(5-HT1) = ¼ 对5-HT2, 5-HT3,多巴胺和肾上腺能受体无作用
5-HT3受体阻断剂 Clebopride Dopamine blocker 胃动力不变 5-HT3 blocker > clebopride 300 folds 无dopamine 拮抗 Metoclopramide Block dopamine & 5-HT3 止吐,胃动力药 granisetron palonosetron Ondansetron Setrons: 治疗肿瘤化疗引起的恶心呕吐
血管紧张素转化酶(angiotensin-converting enzyme, ACE)抑制剂 Angiotensin I Bradykinin: 血管舒张剂 Asp-Arg-Val-Tyr-Ile-His-Pro-Phe-His-Leu Arg-Pro-Pro-Gly-Phe-Ser-Pro-Phe-Arg ACE ACE Asp-Arg-Val-Tyr-Ile-His-Pro-Phe Arg-Pro-Pro-Gly-Phe-Ser-Pro (angiotensin II: 强效收缩血管)
ACE小分子拟肽抑制剂 captopril enalapril Enalaprilat (enalapril的代谢活性组分) ACE的功能 羧肽酶A的作用模式 肽类抑制剂的结合模式 依那普利等 卡托普利 羧烷基脯氨酸
H2受体拮抗剂类抗溃疡药 • 选定靶点-组胺H2受体 (确立研发目标-抑制胃酸分泌药物) • 建立动物筛选模型-麻醉兔灌胃 • 从H2受体天然激动剂-组胺入手,以其为先导结构,保留咪唑环,改变侧链,开始优化
IIb/IIIa糖蛋白受体拮抗剂 • 血栓形成的关键步骤是纤维蛋白原与血小板IIb/IIIa受体结合。 • 被IIb/IIIa受体识别和相互作用的主要区段是纤维蛋白原的三肽片断Arg-Gly-Asp(RGD)。 • 蛇毒或水蛭素中含有RGD的线形或环状肽,是阻断IIb/IIIa受体活化从而抑制血小板聚集的药效团。 • 含有或模拟RGD结构的肽或拟肽可作为纤维蛋白原的拮抗剂,是创制抗血栓药物的一个新途径。 RGD Sibrofiban Roxifiban
4、SOSA 磺胺治疗细菌感染时,发现利尿作用. Acetazolamide 口服利尿 hydrochlorthiazide Dichlorphenamide chlorthiazide bendroflumethiazide 长效利尿药 cyclothiazide 长效利尿药
Sulfasomizole治疗伤寒病,胰岛素释放急性和持久性低血糖Sulfasomizole治疗伤寒病,胰岛素释放急性和持久性低血糖 sulfasomizole Carbutamide, 更强的降糖作用,治疗糖尿病 Iproniazid治疗结核病,情绪提高 抑制单胺氧化酶MAO靶标抑郁型精神病 phenelzine Isoniazid 无抗抑郁 iproniazid 抗抑郁药 tranylcypromine selegiline
Zaprinast sildenafil cGMP:扩张血管,增加血流量 cGMPNO第二信使 增强勃起功能(ED) 抗过敏药扩张血管、降血压抑制PDE5:治疗心绞痛 模拟创新:
5、基于代谢作用 • 代谢活化 • 活性代谢物作为先导物 • 前药设计 • 代谢失活 • 软药设计
6、幸运 结构解析错误
More good luck: Penicillin Chlordiazepoxide (Roche) cisplatin carboplatin oxaliplatin lobaplatin
7、Fragment based lead discovery Fragment-based Screening Fragments: 102 ~104 RO3 compounds Fragment grow, link and merge Target Full screening Lipinski's RO5 Library Synthesis High Throughput Screening (~106 compounds, ~$$2M/Screening) Small is better (sample bigger chemical space, higher hit rate, higher Ligand Efficiency, less false positive results, more intuitive to medicinal chemists)
c a b Biacore Validation HSQC NMR Competitive Binding Experiment Biacore Evolution Fragment Based Lead Discovery Target X-ray / NMR Structures Fragment Library *Hubbard et al (2007), Curr Topics Med Chem, 7, 1568
Weak binding between the target and a small molecule fragment is detected by biophysical methods, e.g., NMR, SPR or cross-validated by these two techniques. Ref: Expert Opin. Drug Discov. (2009) 4:1125
Fragment Selection RO3: 110 < MW <250 ~ 300 cLogP < 2 ~ 3 (or cLogD < 2 ~ 3) 2 < N+O < 6 logS > -4.5 TPSA < 110 Maximize the shape and electrical diversities (Openbabel + Pipeline Pilot) to cover the chemical space for prototypical compounds High ligand efficiency LE = RTln(KD)/HAC LE improves from ~0.3 to ~0.5 during hits to leads (H2L) Final: 1K~2K fragment library (Commercial library: Maybridge or Chembridge)
Most used Fragments Screening methods >10 fragment derived compounds in clinical phase II from pharmaceutical companies, e.g., Novartis, Lily, Abbott, Genentech, Astex, Vernalis, deCODE, Plexxikon. >20 fragment derived compounds in clinical phase I since 2005.
Evolving Fragments - In Practice Growing AT9283 Phase II 0.07 mM Fragment 1 mM 0.003 mM Linking ABT-263 Phase II A B Merging Known Ligands Virtual Screen NVP-AUY922 Phase I/II Detailed Design
药物化学总论(2010,p609) “在发现苗头和先导物的早期研究阶段,同时关注分子的物化性质和活性,以降低新药研发链中各个环节上的风险率,已经成为业界之共识。基于片段的药物设计,辅以配体效率的“监督”,已在国外许多药厂和大学蓬勃展开,大有超过高通量筛选的势头。应用FBDD的13年来,已有一批候选药物进入临床试验阶段。这种研发策略,涉及了分子药理学、结构生物学、分子模拟、化学合成和药物化学等多种学科和技术,整合性很强。在我国,尚未发现采用该方法的研究报道。本节之目的在于呼唤和催生FBDD在我国的实施。”






















