Download
1 / 1
10 likes | 73 Views
Funded by the Wellcome Trust. Dynamical behaviour of water in nanopores by computer simulations Oliver Beckstein* and Mark S. P. Sansom. Laboratory of Molecular Biophysics, Department of Biochemistry, University of Oxford, South Parks Rd, Oxford OX1 3QU, U.K. *e-mail: oliver@biop.ox.ac.uk.

E N D
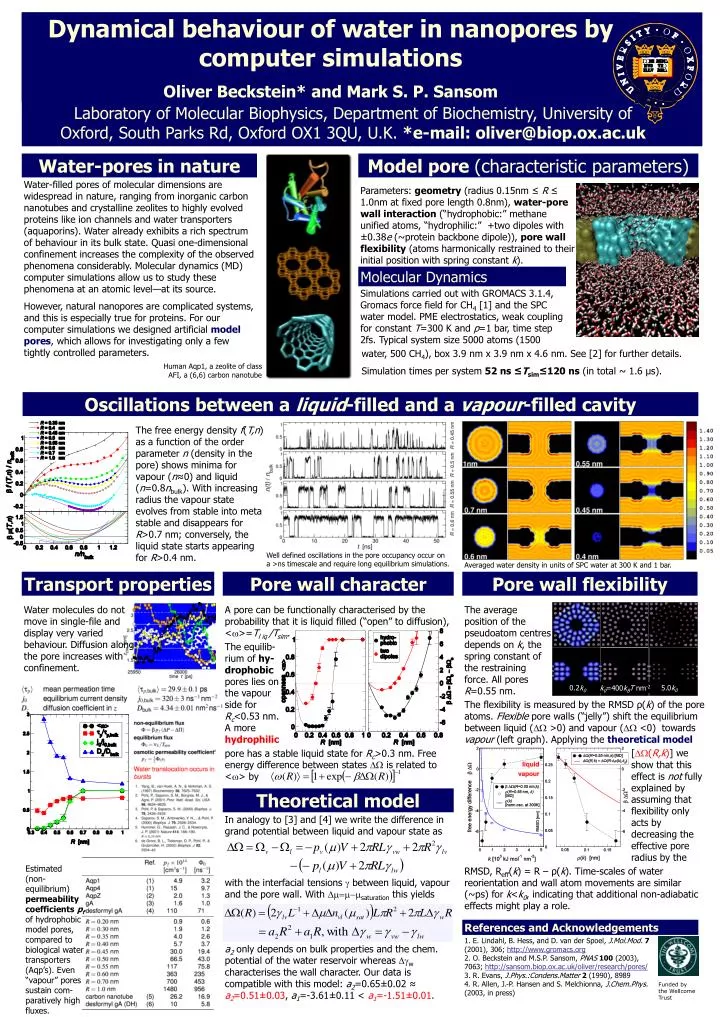
Funded by the Wellcome Trust Dynamical behaviour of water in nanopores by computer simulations Oliver Beckstein* and Mark S. P. Sansom Laboratory of Molecular Biophysics, Department of Biochemistry, University of Oxford, South Parks Rd, Oxford OX1 3QU, U.K. *e-mail: oliver@biop.ox.ac.uk Water-pores in nature Model pore (characteristic parameters) Water-filled pores of molecular dimensions are widespread in nature, ranging from inorganic carbon nanotubes and crystalline zeolites to highly evolved proteins like ion channels and water transporters (aquaporins). Water already exhibits a rich spectrum of behaviour in its bulk state. Quasi one-dimensional confinement increases the complexity of the observed phenomena considerably. Molecular dynamics (MD) computer simulations allow us to study these phenomena at an atomic level—at its source. However, natural nanopores are complicated systems, and this is especially true for proteins. For our computer simulations we designed artificial model pores, which allows for investigating only a few tightly controlled parameters. Parameters: geometry (radius 0.15nm ≤ R ≤ 1.0nm at fixed pore length 0.8nm), water-pore wall interaction (“hydrophobic:” methane unified atoms, “hydrophilic:” +two dipoles with ±0.38e (~protein backbone dipole)), pore wall flexibility (atoms harmonically restrained to their initial position with spring constant k). Molecular Dynamics Simulations carried out with GROMACS 3.1.4, Gromacs force field for CH4 [1] and the SPC water model. PME electrostatics, weak coupling for constant T=300 K and p=1 bar, time step 2fs. Typical system size 5000 atoms (1500 water, 500 CH4), box 3.9 nm x 3.9 nm x 4.6 nm. See [2] for further details. Simulationtimes per system 52 ns ≤Tsim≤120 ns (in total ~ 1.6 μs). Human Aqp1, a zeolite of class AFI, a (6,6) carbon nanotube Oscillations between a liquid-filled and a vapour-filled cavity The free energy density f(T,n) as a function of the order parameter n (density in the pore) shows minima for vapour (n≈0) and liquid (n=0.8nbulk). With increasing radius the vapour state evolves from stable into meta stable and disappears for R>0.7 nm; conversely, the liquid state starts appearing for R>0.4 nm. Well defined oscillations in the pore occupancy occur on a >ns timescale and require long equilibrium simulations. Averaged water density in units of SPC water at 300 K and 1 bar. Transport properties Pore wall character Pore wall flexibility Water molecules do not move in single-file and display very varied behaviour. Diffusion along the pore increases with confinement. A pore can be functionally characterised by the probability that it is liquid filled (“open” to diffusion), <>=Tl iq /Tsim. The average position of the pseudoatom centres depends on k, the spring constant of the restraining force. All pores R=0.55 nm. The equilib-rium of hy-drophobic pores lies on the vapour side for Rc<0.53 nm. A more hydrophilic 5.0k0 0.2k0 k0=400kBT nm-2 The flexibility is measured by the RMSD ρ(k) of the pore atoms. Flexible pore walls (“jelly”) shift the equilibrium between liquid (>0) and vapour (<0) towards vapour (left graph). Applying the theoretical model [(R,k)] we show that this effect is not fully explained by assuming that flexibility only acts by decreasing the effective pore radius by the pore has a stable liquid state for Rc>0.3 nm. Free energy difference between states is related to <> by Theoretical model In analogy to [3] and [4] we write the difference in grand potential between liquid and vapour state as Estimated (non-equilibrium) permeability coefficients pf of hydrophobic model pores, compared to biological water transporters (Aqp’s). Even “vapour” pores sustain com-paratively high fluxes. RMSD, Reff(k) = R– ρ(k). Time-scales of water reorientation and wall atom movements are similar (~ps) for k<k0, indicating that additional non-adiabatic effects might play a role. with the interfacial tensions between liquid, vapour and the pore wall. With Dm=m-msaturation this yields References and Acknowledgements 1. E. Lindahl, B. Hess, and D. van der Spoel, J.Mol.Mod.7 (2001), 306; http://www.gromacs.org 2. O. Beckstein and M.S.P. Sansom, PNAS100 (2003), 7063; http://sansom.biop.ox.ac.uk/oliver/research/pores/ 3. R. Evans, J.Phys.:Condens.Matter2 (1990), 8989 4. R. Allen, J.-P. Hansen and S. Melchionna, J.Chem.Phys. (2003, in press) a2 only depends on bulk properties and the chem. potential of the water reservoir whereas w characterises the wall character. Our data is compatible with this model: a2=0.65±0.02 ≈ a2=0.51±0.03, a1=-3.61±0.11 < a1=-1.51±0.01.