Download

1 / 49
500 likes | 972 Views
Sculptra- P030050. Herbert Lerner, MD Division of General, Restorative and Neurological Devices Plastic and Reconstructive Surgical Devices Branch FDA March 25, 2004. Sculptra- Indication for Use.

E N D
Sculptra- P030050 • Herbert Lerner, MD • Division of General, Restorative and Neurological Devices • Plastic and Reconstructive Surgical Devices Branch • FDA • March 25, 2004
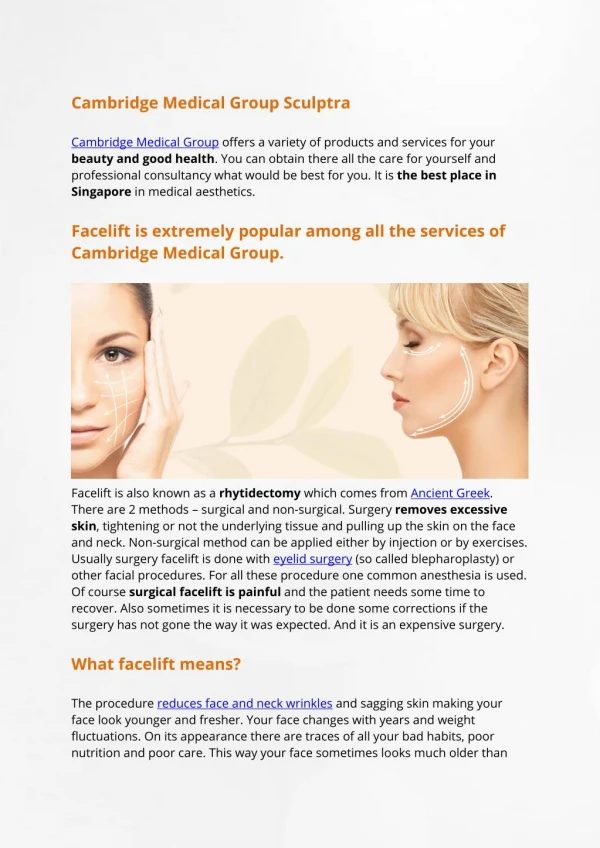
Sculptra- Indication for Use • Sculptra is intended to correct shape and contour deficiencies resulting from facial fat loss (lipoatrophy) in people with human immunodeficiency virus.
Material • Sculptra is a sterile solution consisting of: • PLLA • Sodium Carboxy-methyl cellulose • Mannitol • Sterile Water
SCULPTRAP030050 • FDA Review Team • Herb Lerner, MD- Clinical & Lead • Charles Durfor, PhD- Pre-clinical • David Berkowitz, VMD- Toxicology • Phyllis Silverman, MS- Statistics • Kim Struble- PharmD- Clinical (CDER) • Sybil Wellstood- Manufacturing • Mary Wollerton- Patient Labeling
Sculptra Toxicology Previous Medical uses of Sculptra Components
Sculptra Physical Characteristics • Molecular weight 40 – 50 thousand • PLLA particles irregular shape • 40-63 microns ± • 2 hours required for optimal suspension • Physically chemically and microbiologically stable for 72 hours after suspended.
Sculptra Resorption Kinetics • No weight loss for 24 weeks in phosphate buffer at pH 7.4 at 37 degrees C. • 19% weight loss at 50 degrees C. • Foreign material seen histologically after intradermal implantation for 90 days in rats.
Published In-Vivo Resorption Studies on PLLA • Resorption rate is function of molecular weight, crystallinity, and particle size. • Compact PLLA rods of 95,000 Daltons were implanted subcutaneously in rats. 1 month 19% degraded 3 months 40% degraded 6 months 56% degraded
Sculptra • New-Fill is the name of the device as it is commercially available outside the US. Sculptra is the intended name of the device as it will be marketed in the US. For this review, the use of these names is interchangeable. The components of the two are identical.
SCULPTRASTUDY DESIGN • Presented are 5 investigator-sponsored studies. • 2 studies are from Europe • 3 studies are from the US • All were single center studies • No study was a randomized, controlled, or blinded study as we are used to seeing for a PMA • All were Open label • TCT (Total Cutaneous Thickness)
Vega Study-France • Inclusion Criteria: • HIV+ • Plasma HIV viral load <5000 copies/ml • Current HAART treatment ≥3 months • HAART for at least 3 years • Buccal adipose tissue <2mm
Vega Study-France • Exclusion Criteria: • Cutaneous Kaposi’s Sarcoma of the face • Infection or concurrent herpes labialis • Previous facial fillers within 6 months • Unwilling to meet study follow-up time tables.
Vega Study-France • Study Design • Fifty (50) patients enrolled to study effects if the device over time • 47 patients completed the trial, 2 withdrew at 72 weeks (schedules) and 1 withdrew due to an unrelated event. • Open label, non-randomized, uncontrolled
Vega Study-France • Study Design • Patients were given bi-weekly injections • Safety endpoints designed to look for changes in biological parameters and AE’s • Efficacy endpoints change in TCT
Vega Study-France • Demographics: • Age- (mean) 44.9 ± 6.8 • Gender- 98% male • Race- 84% Caucasian 6% Hispanic 4% North African 2% Black African 4% Carribean
Vega Study-France • Demographics: • AIDS defining event- 50% • CD4 count- 397.1± 168 • HIV viral load-(median)- 200 copies/ml (50-96k) (viral load <5000 copies/ml- 86% of pts.) • TCT cheeks- mean 3.0mm • Adipose tissue <2mm
Vega Study-France • Endpoints: • Safety- adverse events • Treatment-related events • Local and systemic • Change from baseline CD4 cells • Viral load • Blood Lactic Acid levels
Vega Study-France • Results • Bruising- 3% • Hematoma- 30% • Nodule- 52%
Vega Study-France • Efficacy- Change from baseline in TCT (mm) • Study demonstrated statistically significant increases from baseline to week 96- • At 8 weeks mean change 5.2 mm (SD 1.7) • At 24 weeks change was 6.4 mm (SD 1.6) • At 48 weeks, change was 7.2mm (SD 1.3) • At 72 weeks, change was 7.2 mm (SD 1.3) • At 96 weeks, change was 7.0mm (SD 1.4) • Photographic Assessment • Visual Analogue Scale 0-10 scale (with 10 the most satisfying physical/emotional state)
Vega Study-France • Figure 1: Profile of Dermal Thickness by Ultrasound by Weeks From First Injection
Chelsea & Westminster- England • Inclusion Criteria: • HIV+ • Moderate to severe lipoatrophy • Not pregnant or lactating
Chelsea & Westminster- England • Exclusion Criteria • Active opportunistic disease or wasting • Current growth hormone therapy • Current chemotherapy for malignancy • Known hypersensitivity to PLLA
Chelsea & Westminster- England • Study Design • 30 patients • Half of group delayed 12 weeks as a comparator • 30 pts. Treated • 29 pts. Reported (1 declined data disclosure)
Chelsea & Westminster- England • Study design • Two groups of 15, the second group had injections delayed for 12 weeks • Clinical exam, serum CD4 and viral loads obtained • Facial Ultrasound • VAS and HAD (Anxiety/ Depression scores)
Chelsea & Westminster- England • Demographics: • Age- 41 years (mean) • Gender- 28 males/ 2 females • Race- 72% Caucasian 3% Black 24% Hispanic
Chelsea & Westminster- England • Demographics: • Mead duration of HAART- 5.1 years • Mean baseline CD4 count- 473.6 • Viral load (median) - 72.0 copies/ml
Chelsea & Westminster- England • Endpoints: • Safety- • Change in viral load • Change in CD4 count • Change in blood chemistry • Adverse Events • Efficacy- • Buccal skin thickness measurements • Change in facial appearance- MD and Pt. assessments
Chelsea & Westminster • Adverse events-combined groups • Injection site bruising- 38% • Injection site discomfort- 10% • Injection site erythema- 10% • Injection site inflammation- 10% • Injection site nodule- 31% • VAS scores improved • Clinical lab parameters unchanged
Chelsea & Westminster • BASELINE TO WEEK 12
Common Finding In Both Studies- • Nodule at injection site • 52% VEGA • 31% C&W • Discussion- • Onset average up to 218 days (9 to 748) • Most reported as mild and not visible • No histological data available
US Studies • APEX-001 • Investigator Sponsored Compassionate Use Study • Open label, uncontrolled, non-randomized study • 100 patients • 1-6 treatment sessions (average-3) • 1-8 cc of New-Fill per treatment session • Demographics similar to previous studies • HIV+ 14 years • Mean age 44.5 • 82/96 patients Caucasian
US Studies • Inclusion Criteria • HIV+ • Demonstrable photographic lipoatrophy • Exclusion Criteria • Active Infection, Kaposi’s sarcoma or Herpes on the face • Facial injections within last 3 months • Treatment with interferon or steroids
US Studies • Safety results- • Adverse events considered mild • 6 nodules reported in 85 patients at 3 weeks • 39 nodules in 70 patients seen at 12 months • Efficacy results- • High patient satisfaction- 8.8/10 • High investigator rating – from 3.2 to 1.36 (lower score is better)
US Studies • APEX002 • Investigator sponsored IDE • HIV+ • 100 patients • Average of 3.5 treatments/patient • Similar demographics • Average time HIV+ 11.9 yrs • Average years HAART therapy- 13
US Studies • Adverse Events- mild • Soreness and nodules • 6 nodules in 99 patients • 19 pts. With injection site soreness • 2 pts. With transient fever • High patient satisfaction- • Scores went from 3.71 to under 1 (lower score is better)
US Studies • US Study- Hermosa Beach • Open label, uncontrolled, non-randomized • Similar demographics and treatment schedule • 1-6 treatments/patient • Up to 6 cc per treatment • Average time HIV+ 13 years • Average time HAART- 9 years
US Studies • Inclusion/Exclusion criteria • Similar to Apex studies • HIV+ • Lipoatrophy • Infections of face, Kaposi's sarcoma • Treatment with interferon or steroids • Uncontrolled DM, lactic acidosis
US Studies • Endpoints • To evaluate the quantifiable improvement in facial wasting after serial intradermal injections of New-Fill • Safety- • In repeated treatments • Efficacy • Durability of New-Fill • Psychological impact on patients
US Studies • Results • Adverse events-mild • 8 nodules in 87 patients • High patient satisfaction • Average increase TCT- 6mm @ 6 mos. • Average mm initial 7.44 • Average mm-end of tx. 13.92 • Average mm- 6 mos. 13.22 • Average mm- increase 5.78
Conclusion- Overall Safety • In general, the majority of treatment related events are mild pain, bruising and swelling at the injection site. • Device events are generally palpable subcutaneous nodules (up to 50%) • No major AE’s reported
Conclusion- Overall Efficacy • TCT analysis in VEGA study demonstrates increased TCT. • Dermal thickness changes in the C&W study also demonstrate significant enhancement of dermal thickness • Photographic evidence of sustained efficacy is shown
Conclusion- Overall Efficacy • Quality of Life assessments show improvement from the baseline
Statistical Summary • No masking or use of validated severity scale • Total Skin Thickness (TCT) was used as surrogate endpoint for improved appearance • Statistically significant (p<0.001) increases in TCT observed in Vega and C&W Studies • Treatment effect was independent of time on HAART, baseline CD4 counts, or baseline skin thickness • Increase in skin thickness correlated pictorially with improved appearance
Panel Question-1 • 21 CFR 860.7(d)(1) states that there is a reasonable assurance that the device is safe when it can be determined that the probable benefits to health from use of the device for its intended uses, when accompanied by adequate instructions for use and warnings against unsafe use, outweigh any probable risks. Considering the data in the PMA, please comment on whether there is a reasonable assurance that the device is safe.
Panel Question- 2 • 21 CFR 860.7(e)(1) states that there is a reasonable assurance that a device is effective when it can be determined, based on valid scientific evidence, that in a significant portion of the target population, the use of the device for its intended uses and conditions of use, when accompanied by adequate directions for use and warnings against unsafe use, will produce clinically significant results. Considering the data in the PMA, is there reasonable assurance that the device is effective?
Panel Question- 3 • Patients in the European studies (79) were followed-up for periods ranging from 24 weeks to 2 years, and those in the U.S. studies (286) were followed up to 2 years. If you agree that there is enough evidence in the PMA to support the safety and effectiveness of the device, do you feel that a post-approval study to assess the long term use of this device should be initiated, and if so, please advise FDA as to the type of data you feel should be collected, and the appropriate duration of follow-up.
Panel Question- 4 • A large volume of this device (up to 11cc. per treatment) is required to achieve an optimal cosmetic effect, and precise placement of the material in the correct dermal plane (deep dermis or subcuticular layer) is important. Please advise FDA whether a physician training program is indicated for those wishing to use this device, and if so, what type of training would be appropriate.