Download
1 / 36
900 likes | 4.86k Views
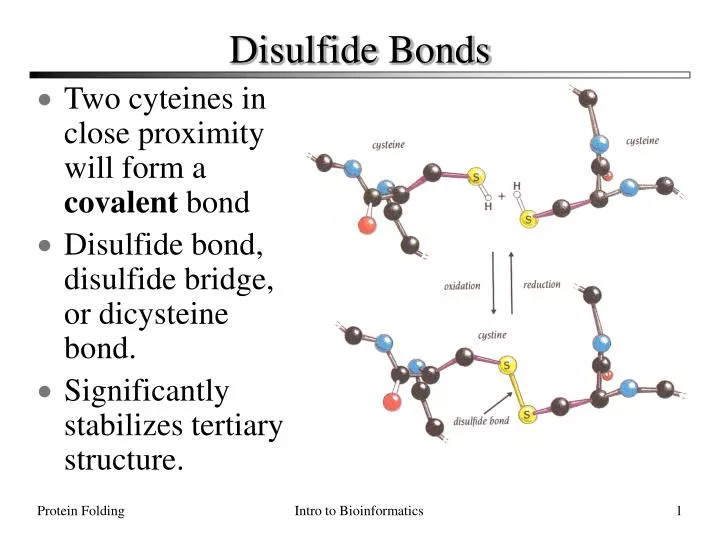
Disulfide Bonds. Two cyteines in close proximity will form a covalent bond Disulfide bond, disulfide bridge, or dicysteine bond. Significantly stabilizes tertiary structure. Determining Protein Structure. There are O(100,000) distinct proteins in the human proteome.

E N D
Disulfide Bonds • Two cyteines in close proximity will form a covalent bond • Disulfide bond, disulfide bridge, or dicysteine bond. • Significantly stabilizes tertiary structure. Protein Folding
Determining Protein Structure • There are O(100,000) distinct proteins in the human proteome. • 3D structures have been determined for 14,000 proteins, from all organisms • Includes duplicates with different ligands bound, etc. • Coordinates are determined by X-ray crystallography Protein Folding
~0.5mm X-Ray Crystallography • The crystal is a mosaic of millions of copies of the protein. • As much as 70% is solvent (water)! • May take months (and a “green” thumb) to grow. Protein Folding
X-Ray diffraction • Image is averagedover: • Space (many copies) • Time (of the diffractionexperiment) Protein Folding
Electron Density Maps • Resolution is dependent on the quality/regularity of the crystal • R-factor is a measure of “leftover” electron density • Solvent fitting • Refinement Protein Folding
The Protein Data Bank • http://www.rcsb.org/pdb/ ATOM 1 N ALA E 1 22.382 47.782 112.975 1.00 24.09 3APR 213 ATOM 2 CA ALA E 1 22.957 47.648 111.613 1.00 22.40 3APR 214 ATOM 3 C ALA E 1 23.572 46.251 111.545 1.00 21.32 3APR 215 ATOM 4 O ALA E 1 23.948 45.688 112.603 1.00 21.54 3APR 216 ATOM 5 CB ALA E 1 23.932 48.787 111.380 1.00 22.79 3APR 217 ATOM 6 N GLY E 2 23.656 45.723 110.336 1.00 19.17 3APR 218 ATOM 7 CA GLY E 2 24.216 44.393 110.087 1.00 17.35 3APR 219 ATOM 8 C GLY E 2 25.653 44.308 110.579 1.00 16.49 3APR 220 ATOM 9 O GLY E 2 26.258 45.296 110.994 1.00 15.35 3APR 221 ATOM 10 N VAL E 3 26.213 43.110 110.521 1.00 16.21 3APR 222 ATOM 11 CA VAL E 3 27.594 42.879 110.975 1.00 16.02 3APR 223 ATOM 12 C VAL E 3 28.569 43.613 110.055 1.00 15.69 3APR 224 ATOM 13 O VAL E 3 28.429 43.444 108.822 1.00 16.43 3APR 225 ATOM 14 CB VAL E 3 27.834 41.363 110.979 1.00 16.66 3APR 226 ATOM 15 CG1 VAL E 3 29.259 41.013 111.404 1.00 17.35 3APR 227 ATOM 16 CG2 VAL E 3 26.811 40.649 111.850 1.00 17.03 3APR 228 Protein Folding
A Peek at Protein Function • Serine proteases – cleave other proteins • Catalytic Triad: ASP, HIS, SER Protein Folding
Cleaving the peptide bond Protein Folding
Three Serine Proteases • Chymotrypsin – Cleaves the peptide bond on the carboxyl side of aromatic (ring) residues: Trp, Phe, Tyr; and large hydrophobic residues: Met. • Trypsin – Cleaves after Lys (K) or Arg (R) • Positive charge • Elastase – Cleaves after small residues: Gly, Ala, Ser, Cys Protein Folding
Specificity Binding Pocket Protein Folding
The Protein Folding Problem • Central question of molecular biology:“Given a particular sequence of amino acid residues (primary structure), what will the tertiary/quaternary structure of the resulting protein be?” • Input: AAVIKYGCAL…Output: 11, 22…= backbone conformation:(no side chains yet) Protein Folding
Protein Folding – Biological perspective • “Central dogma”: Sequence specifies structure • Denature – to “unfold” a protein back to random coil configuration • -mercaptoethanol – breaks disulfide bonds • Urea or guanidine hydrochloride – denaturant • Also heat or pH • Anfinsen’s experiments • Denatured ribonuclease • Spontaneously regained enzymatic activity • Evidence that it re-folded to native conformation Protein Folding
Folding intermediates • Levinthal’s paradox – Consider a 100 residue protein. If each residue can take only 3 positions, there are 3100 = 5 1047 possible conformations. • If it takes 10-13s to convert from 1 structure to another, exhaustive search would take 1.6 1027 years! • Folding must proceed by progressive stabilization of intermediates • Molten globules – most secondary structure formed, but much less compact than “native” conformation. Protein Folding
Forces driving protein folding • It is believed that hydrophobic collapse is a key driving force for protein folding • Hydrophobic core • Polar surface interacting with solvent • Minimum volume (no cavities) • Disulfide bond formation stabilizes • Hydrogen bonds • Polar and electrostatic interactions Protein Folding
Folding help • Proteins are, in fact, only marginally stable • Native state is typically only 5 to 10 kcal/mole more stable than the unfolded form • Many proteins help in folding • Protein disulfide isomerase – catalyzes shuffling of disulfide bonds • Chaperones – break up aggregates and (in theory) unfold misfolded proteins Protein Folding
The Hydrophobic Core • Hemoglobin A is the protein in red blood cells (erythrocytes) responsible for binding oxygen. • The mutation E6V in the chain places a hydrophobic Val on the surface of hemoglobin • The resulting “sticky patch” causes hemoglobin S to agglutinate (stick together) and form fibers which deform the red blood cell and do not carry oxygen efficiently • Sickle cell anemia was the first identified molecular disease Protein Folding
Sickle Cell Anemia Sequestering hydrophobic residues in the protein core protects proteins from hydrophobic agglutination. Protein Folding
Computational Problems in Protein Folding • Two key questions: • Evaluation – how can we tell a correctly-folded protein from an incorrectly folded protein? • H-bonds, electrostatics, hydrophobic effect, etc. • Derive a function, see how well it does on “real” proteins • Optimization – once we get an evaluation function, can we optimize it? • Simulated annealing/monte carlo • EC • Heuristics • We’ll talk more about these methods later… Protein Folding
Fold Optimization • Simple lattice models (HP-models) • Two types of residues: hydrophobic and polar • 2-D or 3-D lattice • The only force is hydrophobic collapse • Score = number of HH contacts Protein Folding
Scoring Lattice Models • H/P model scoring: count noncovalent hydrophobic interactions. • Sometimes: • Penalize for buried polar or surface hydrophobic residues Protein Folding
What can we do with lattice models? • For smaller polypeptides, exhaustive search can be used • Looking at the “best” fold, even in such a simple model, can teach us interesting things about the protein folding process • For larger chains, other optimization and search methods must be used • Greedy, branch and bound • Evolutionary computing, simulated annealing • Graph theoretical methods Protein Folding
Learning from Lattice Models • The “hydrophobic zipper” effect: Ken Dill ~ 1997 Protein Folding
Representing a lattice model • Absolute directions • UURRDLDRRU • Relative directions • LFRFRRLLFFL • Advantage, we can’t have UD or RL in absolute • Only three directions: LRF • What about bumps? LFRRR • Bad score • Use a better representation Protein Folding
Preference-order representation • Each position has two “preferences” • If it can’t have either of the two, it will take the “least favorite” path if possible • Example: {LR},{FL},{RL},{FR},{RL},{RL},{FR},{RF} • Can still cause bumps:{LF},{FR},{RL},{FL},{RL},{FL},{RF},{RL},{FL} Protein Folding
“Decoding” the representation • The optimizer works on the representation, but to score, we have to “decode” into a structure that lets us check for bumps and score. • Example: How many bumps in: URDDLLDRURU? • We can do it on graph paper • Start at 0,0 • Fill in the graph • In PERL we use a two-dimensional array Protein Folding
A two-dimensional array in PERL $configuration = “URDDLLDRURU”; $sequence = “HPPHHPHPHHH”; foreach $i (1..100) { foreach $j (1..100) { $grid[$i][$j] = “empty”; } } $x = 0; $y = 0; @moves = split(//,$configuration); @residues = split(//,$sequence); Protein Folding
Setting up the grid foreach $move (@moves) { $residue = shift(@residues); if ($move = “U”) { $y_position++; } if ($move = “R”) { $x_position++; } etc… if ($grid[$x][$y] ne “empty”) { BUMP! } else { $grid[$x][$y] = $residue; } Protein Folding
More realistic models • Higher resolution lattices (45° lattice, etc.) • Off-lattice models • Local moves • Optimization/search methods and / representations • Greedy search • Branch and bound • EC, Monte Carlo, simulated annealing, etc. Protein Folding
The Other Half of the Picture • Now that we have a more realistic off-lattice model, we need a better energy function to evaluate a conformation (fold). • Theoretical force field: • G = Gvan der Waals + Gh-bonds + Gsolvent + Gcoulomb • Empirical force fields • Start with a database • Look at neighboring residues – similar to known protein folds? Protein Folding
Threading: Fold recognition • Given: • Sequence: IVACIVSTEYDVMKAAR… • A database of molecular coordinates • Map the sequence onto each fold • Evaluate • Objective 1: improve scoring function • Objective 2: folding Protein Folding
Secondary Structure Prediction AGVGTVPMTAYGNDIQYYGQVT… A-VGIVPM-AYGQDIQY-GQVT… AG-GIIP--AYGNELQ--GQVT… AGVCTVPMTA---ELQYYG--T… AGVGTVPMTAYGNDIQYYGQVT… ----hhhHHHHHHhhh--eeEE… Protein Folding
Secondary Structure Prediction • Easier than folding • Current algorithms can prediction secondary structure with 70-80% accuracy • Chou, P.Y. & Fasman, G.D. (1974). Biochemistry, 13, 211-222. • Based on frequencies of occurrence of residues in helices and sheets • PhD – Neural network based • Uses a multiple sequence alignment • Rost & Sander, Proteins, 1994 , 19, 55-72 Protein Folding
Chou-Fasman Parameters Protein Folding
Chou-Fasman Algorithm • Identify -helices • 4 out of 6 contiguous amino acids that have P(a) > 100 • Extend the region until 4 amino acids with P(a) < 100 found • Compute P(a) and P(b); If the region is >5 residues and P(a) > P(b) identify as a helix • Repeat for -sheets [use P(b)] • If an and a region overlap, the overlapping region is predicted according to P(a) and P(b) Protein Folding
Chou-Fasman, cont’d • Identify hairpin turns: • P(t) = f(i) of the residue f(i+1) of the next residue f(i+2) of the following residue f(i+3) of the residue at position (i+3) • Predict a hairpin turn starting at positions where: • P(t) > 0.000075 • The average P(turn) for the four residues > 100 • P(a) < P(turn) > P(b) for the four residues • Accuracy 60-65% Protein Folding
Chou-Fasman Example • CAENKLDHVRGPTCILFMTWYNDGP • CAENKL – Potential helix (!C and !N) • Residues with P(a) < 100: RNCGPSTY • Extend: When we reach RGPT, we must stop • CAENKLDHV: P(a) = 972, P(b) = 843 • Declare alpha helix • Identifying a hairpin turn • VRGP: P(t) = 0.000085 • Average P(turn) = 113.25 • Avg P(a) = 79.5, Avg P(b) = 98.25 Protein Folding