Download

1 / 30
390 likes | 542 Views
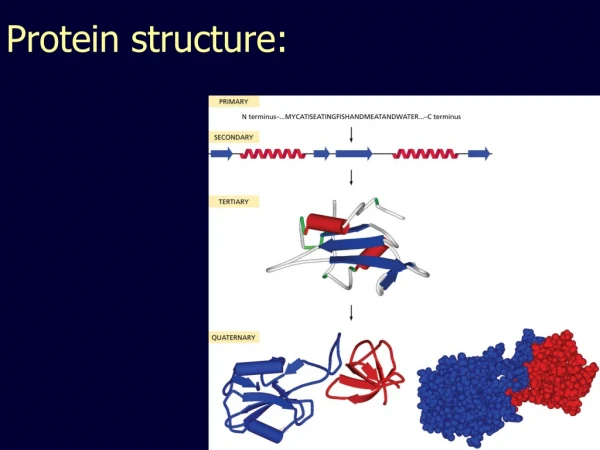
Protein Structure. Tertiary structure determination Evaluation Comparison. Supplemental primer on protein structure: http://www.ncbi.nlm.nih.gov/About/primer/molecularmod.html. Reading: Chapter 13. BIO520 Bioinformatics Jim Lund. X-ray crystallography

E N D
Protein Structure Tertiary structure determination Evaluation Comparison Supplemental primer on protein structure: http://www.ncbi.nlm.nih.gov/About/primer/molecularmod.html Reading: Chapter 13 BIO520 Bioinformatics Jim Lund
X-ray crystallography Nobel prizes: Perutz and Kendrew,1962 (myoglobin) MacKinnon 2002 (ion channels) Kornberg, 2006 (ribosome) requires crystals NMR methods Nobel prize 2002, Kurt Wüthrich <30kD Protein Structure3D Determination
Purify protein Crystallize protein Test an array of conditions to determine which give good quality crystals. X-ray diffraction Compute atom locations (C, N, O, S) Relax structure computationally (energy minimization) X-ray crystallography
Isotopically label protein NMR: NOEs, J-coupling constants measurements give structural constraints Compute structures using constraints. Relax structure computationally (energy minimization) NMR Structure Determination
Local environment of an atom alters the emitted radio frequency The exact B field that a given nucleus “feels” is dependent on the shielding(s): - the chemical nature of bonded atoms - the dihedral angle - the presence of other local fields (ring currents in aromatic groups) - paramagnetic centers (unpaired e-) - the chemical shift (in Hz) depends on Bo and is small compared to the Larmour freq.
Protein Structure Elucidation via NMR Structure Calculation – simulated annealing Fold up random conformation to minimize violations of constraints.
Degree of folding (folded, partially folded, unfolded) and kind of fold (primarily alpha-helix or beta-sheet) of a protein independent from a biological test of activity. Optimization of conditions such as pH, temperature, salt concentration etc. for best solubility, folding, least aggregation etc in short time. With 15N-labelling (not radioactive!) counting of folded/unfolded residues becomes possible; this might help to determine domain borders more accurately when cloning protein domains. Binding studies are possible with and without isotope labeling. Ligand conformations can be determined. Reaction products or intermediates can be investigated. Folding and unfolding studies for slow kinetics (ms - sec - min) are possible, e.g. with H/D-exchange experiments or stopped-flow deuterium-pulse-experiments. Dynamics-investigations yield information on the degree of oligomerization (monomer/dimer/...) as well as mobility of active sites of the protein. Applications of NMR
Torsion angles in disallowed Ramachandran plot regions Maximize H-bonding Minimize exposed hydrophobic side chains Maximize exposed polar/charged side chains Holes->bad Minimize distorted H-bonds, distorted helix angles Low R-factor (X-ray structure), low backbone RMSD (NMR) Structure Evaluation
DSSP Identify secondary struct, solvent accessibility (buried or surface aa) PROCHECK Calculates torsion angles, bond angles, interatomic distances VADAR (Volume, Area, Dihedral Angle Reporter) Classifies secondary struct, salt bridges Solvent accessibility (buried or surface aa), buried charge Structure Evaluation Programs
Homology modeling SwissModel, SDSC1, CPH Models Threading SAMt99, 3D-PSSM, FUGUE Ab initio GAMESS, Rosetta (Baker lab) EVA: Evaluation of automatic structure prediction servers (http://cubic.bioc.columbia.edu/eva/) Protein Structure Prediction
Align query protein to known structure. Replace known with query. Backbone insertions/deletions. Side chains. Refine model using energy minimization. SwissModel, SDSC1, CPH Models Homology modeling
Align sequences, one has known structure “thread” backbone of test sequence into known structure. minimize energies predicts 3D structure Sequence of unknown structure “thread” through all possible folds Threading (Fold Recognition)
Generate a .pdb from a thread of a >30% similar homolog http://www.expasy.ch/swissmod/SWISS-MODEL.html Search for “threads” in proteins 0-25% similar to your query (structural analogs without homology) http://www.predictprotein.org/ Threading Services
Practical, rapid Apply to many problems thousands of atoms different atom types proteins, organics, nucleic acids, drugs dynamic: kinetics and thermodynamics interactions DOCKING Molecular Mechanics
Two components: Devising a scoring (ie, energy) function that can distinguish between correct (native or native-like) structures from incorrect ones. A search method to explore the conformational space. Ab initio prediction
Nuclei, electrons lumped into atoms All atoms are spherical Atoms interact on springs, via classical potentials Interactions determine spatial distribution determine energies of states Molecular Mechanics Assumptions
Energy Calculations Image: http://cmm.info.nih.gov/intro_simulation/
Energy Energy = Stretch+Bend+Torsion+Non-bonded Charge van der Waals Dipole...
“Simple” Calculations E=k(r-r0)2 Bond stretching
Repulsion Depends on Attraction Charge Non-bonded Energies E= -Aij+Bij+ qiqj i j rij6 rij12 i j rij
Software packages for molecular dynamics: SYBL AMBER CHARMM Geometry/Computation Used for: Ab initio Energy Minimization Docking
Single-site mutation Effect on protein active site, eg Energy Minimization of Residue Conformers Molecular mechanics: “Tweaking” ESyPred3D "Poor Man's Version"
Energy minimize bound molecules combinatorial optimization Important in drug design/modelling Docking QSAR/Superposition Combinatorial Chemistry
RELIWE http://bioinf.mpi-inf.mpg.de/reliwe/reliwe/reliwe_home.html DOCK http://dock.compbio.ucsf.edu/ AutoDock http://autodock.scripps.edu/ Docking Programs
Recognize protein 3D similarities w/o sequence similarity restricted to known 3D Heirarchical databases CATH Class(C), Architecture(A), Topology(T) and Homologous superfamily (H) Computational SCOP Structural Classification of Proteins hand annotated Structure similarity searches Structure Neighboring
Similar groups of secondary structure DALI Distance matrices CE, VAST (Vector Alignment Search Tool) Vector representation of secondary structure VAST Identify pairs of secondary structure elements with similar type, orientation, connection, then extend these matches. NCBI tool Can view aligned structures with Cn3D Structure comparison searches