Download
1 / 37
720 likes | 1.8k Views
Genome Annotation. Rosana O. Babu. Sequence to Annotation. Input1-Variant Annotation. Input2- Structural Annotation. Structural Annotation was conducted using AUGUSTUS (version 2.5.5), Magnaporthe_grisea as genome model

E N D
Genome Annotation Rosana O. Babu
Input2- Structural Annotation • Structural Annotation was conducted using AUGUSTUS (version 2.5.5), Magnaporthe_grisea as genome model • However, we have to develop genome model for Oomycete to obtain accurate result
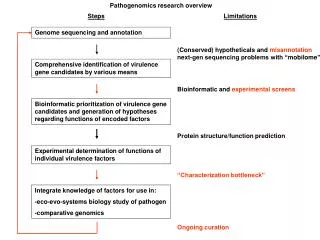
Genome Annotation • The process of identifying the locations of genes and the coding regions in a genome to determe what those genes do • Finding and attaching the structural elements and its related function to each genome locations
Genome Annotation gene function prediction Attaching biological information to these elements- eg: for which protein exon will code for gene structure prediction Identifying elements (Introns/exons,CDS,stop,start) in the genome
Eukaryote genome annotation Find locus Genome Transcription Primary Transcript RNA processing Find exons using transcripts ATG STOP Processed mRNA m7G AAAn Translation Find exons using peptides Polypeptide Protein folding Folded protein Find function Enzyme activity A B Functional activity
Prokaryote genome annotation Find locus Genome Transcription Primary Transcript RNA processing Find CDS START STOP START STOP Processed RNA Translation Polypeptide Protein folding Folded protein Find function Enzyme activity A B Functional activity
Genome annotation - workflow Genome sequence Masked or un-masked genome sequence Repeats Structural annotation-Gene finding nc-RNAs, Introns Protein-coding genes Functional annotation Viewed & Released in Genome viewer
Genome Repeats & features Polymorphic between individuals/populations • Percentage of repetitive sequences in different organisms • Microsatellite • Minisatellite • Tandem repeat • Short tandem repeat • SSR
Finding repeats as a preliminary to gene prediction • Repeat discovery • Literature and public databanks • Homology based approaches • Automated approaches (e.g. RepeatScout or RECON) • Tandem repeats: Tandem, TRF • Use RepeatMasker to search the genome and mask the sequence
Masked sequence • Repeatmasked sequence is an artificial construction where those regions which are thought to be repetitive are marked with X’s • Widely used to reduce the overhead of subsequent computational analyses and to reduce the impact of TE’s in the final annotation set >my sequence atgagcttcgatagcgatcagctagcgatcaggctactattggcttctctagactcgtctatctctattagctatcatctcgatagcgatcagctagcgatcaggctactattggcttcgatagcgatcagctagcgatcaggctactattggcttcgatagcgatcagctagcgatcaggctactattggctgatcttaggtcttctgatcttct >my sequence (repeatmasked) atgagcttcgatagcgatcagctagcgatcaggctactattxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxatctcgatagcgatcagctagcgatcaggctactattxxxxxxxxxxxxxxxxxxxtagcgatcaggctactattggcttcgatagcgatcagctagcgatcaggctxxxxxxxxxxxxxxxxxxxtcttctgatcttct Positions/locations are not affected by masking
Types of Masking- Hard or Soft? • Sometimes we want to mark up repetitive sequence but not to exclude it from downstream analyses. This is achieved using a format known as soft-masked >my sequence ATGAGCTTCGATAGCGCATCAGCTAGCGATCAGGCTACTATTGGCTTCTCTAGACTCGTCTATCTCTATTAGTATCATCTCGATAGCGATCAGCTAGCGATCAGGCTACTATTGGCTTCGATAGCGATCAGCTAGCGATCAGGCTACTATTGGCTTCGATAGCGATCAGCTAGCGATCAGGCTACTATTGGCTGATCTTAGGTCTTCTGATCTTCT >my sequence (softmasked) ATGAGCTTCGATAGCGCATCAGCTAGCGATCAGGCTACTATTggcttctctagactcgtctatctctattagtatcATCTCGATAGCGATCAGCTAGCGATCAGGCTACTATTggcttcgatagcgatcagcTAGCGATCAGGCTACTATTggcttcgatagcgatcagcTAGCGATCAGGCTACTATTGGCTGATCTTAGGTCTTCTGATCTTCT >my sequence (hardmasked) atgagcttcgatagcgatcagctagcgatcaggctactattxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxxatctcgatagcgatcagctagcgatcaggctactattxxxxxxxxxxxxxxxxxxxtagcgatcaggctactattggcttcgatagcgatcagctagcgatcaggctxxxxxxxxxxxxxxxxxxxtcttctgatcttct
Genome annotation - workflow Genome sequence Masked or un-masked Map repeats Gene finding- structural annotation nc-RNAs, Introns Protein-coding genes Functional annotation Viewed & Released in Genome viewer
Structural annotation Identification of genomic elements • Open reading frame and their localization • Coding regions • Location of regulatory motifs • Start/Stop • Splice Sites • Non coding Regions/RNA’s
Methods • Similarity • Similarity between sequences which does not necessarily infer any evolutionary linkage • Ab- initio prediction • Prediction of gene structure from first principles using only the genome sequence
Genefinding ab initio similarity
Gene_finding resources for Homology based methods • Transcript • cDNA sequences • EST sequences • Peptide • Non-redundant (nr) protein database • Protein sequence data, Mass spectrometry data • Genome • Other genomic sequence
ab initio prediction Genome Coding potential ATG & Stop codons Splice sites ATG & Stop codons Coding potential
Genefinding - ab initio predictions • Use compositional features of the DNA sequence to define coding segments (essentially exons) • ORFs • Coding bias • Splice site consensus sequences • Start and stop codons • Methods • Training sets are required • Each feature is assigned a log likelihood score • Use dynamic programming to find the highest scoring path for accuracy • Examples: Genefinder, Augustus, Glimmer, SNAP, fgenesh
Genefinding - similarity • Use known coding sequence to define coding regions • EST sequences • Peptide sequences • Problem to handle fuzzy alignment regions around splice sites • Examples: EST2Genome, exonerate, genewise Gene-finding - comparative • Use two or more genomic sequences to predict genes based on conservation of exon sequences • Examples: Twinscan and SLAM
Genefinding - non-coding RNA genes • Non-coding RNA genes can be predicted using knowledge of their structure or by similarity with known examples • tRNAscan - uses an HMM and co-variance model for prediction of tRNA genes • Rfam - a suite of HMM’s trained against a large number of different RNA genes
Gene-finding omissions • Alternative isoforms • Currently there is no good method for predicting alternative isoforms • Only created where supporting transcript evidence is present • Pseudogenes • Each genome project has a fuzzy definition of pseudogenes • Badly curated/described across the board • Promoters • Rarely a priority for a genome project • Some algorithms exist but usually not integrated into an annotation set
Practical- structural annotation Eukaryotes- AUGUSTUS (gene model) ~/Programs/augustus.2.5.5/bin/augustus --strand=both --genemodel=partial --singlestrand=true --alternatives-from-evidence=true --alternatives-from-sampling=true --progress=true --gff3=on --uniqueGeneId=true --species=magnaporthe_griseaour_genome.fasta>structural_annotation.gff Prokaryotes – PRODIGAL (Codon Usage table) ~/Programs/prodigal.v2_60.linux -a protein_file.fa -g 11 –d nucleotide_exon_seq.fa -f gff -i contigs.fa -o genes_quality.txt -s genes_score.txt -t genome_training_file.txt
Structural Annotation- • Structural Annotation was conducted using AUGUSTUS (version 2.5.5), Magnaporthe_grisea as genome model • However, we have to develop genome model for obtaining accurate result
Functional annotation Attaching biological information to genomic elements • Biochemical function • Biological function • Involved regulation and interactions • Expression • Utilise known structural information to predicted protein sequence
Genome annotation - workflow Genome sequence Masked or un-masked Map repeats Gene finding- structural annotation nc-RNAs, Introns Protein-coding genes Functional annotation Viewed & Released in Genome viewer
Genome annotation Genome Transcription Primary Transcript RNA processing ATG STOP Processed mRNA m7G AAAn Translation Polypeptide Protein folding Folded protein Find function Enzyme activity A B Functional activity
Functional annotation – Homology Based • Predicted Exons/CDS/ORF are searched against the non-redundant protein database (NCBI, SwissProt) to search for similarities • Visually assess the top 5-10 hits to identify whether these have been assigned a function • Functions are assigned
Functional annotation - Other features • Other features which can be determined • Signal peptides • Transmembrane domains • Low complexity regions • Various binding sites, glycosylation sites etc. • Protein Domain See http://expasy.org/tools/ for a good list of possible prediction algorithms
Functional annotation - Other features (Ontologies) • Use of ontologies to annotate gene products • Gene Ontology (GO) • Cellular component • Molecular function • Biological process
Practical - FUNCTIONAL ANNOTATION • Homology Based Method • setup blast database for nucleotide/protein • Blasting the genome.fasta for annotations (nucleotide/protein) • sorting for blast minimum E-value (>=0.01) for nucleotide/protein • Further filtering for best blast hit (5-15) and assigning functions • Removing Positive strand blast hits • Removing negative strand blast hits
Functional annotation- output Bioinformatics tools for Comparative Genomics of Vectors
Conclusion • Annotation accuracy is only as good as the available supporting data at the time of annotation- update information is necessary • Gene predictions will change over time as new databecomes available (ESTs, related genomes) that are much similar than previous ones • Functional assignments will change over time as new data becomes available (characterization of hypothetical proteins)