Download

1 / 67
820 likes | 1.73k Views
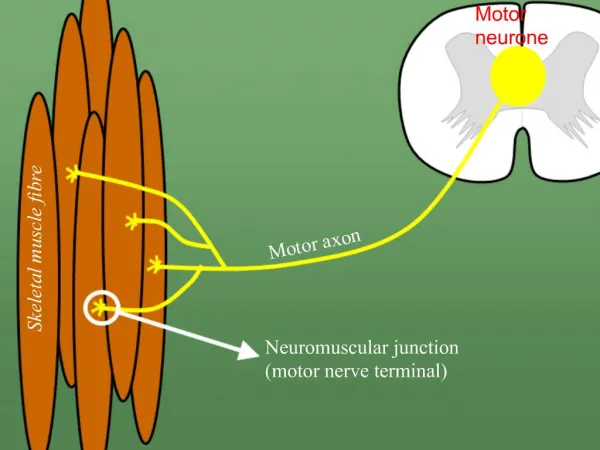
Neuromuscular Junction Disorders. Abdullah Al-Salti R3 AHD 9 march 2011. Neuromuscular junction. Myasthenia Gravis. Myasthenia Gravis: a chronic autoimmune neuromuscular disease characterized by varying degrees of weakness of the skeletal (voluntary) muscles of the body. Age and Gender:

E N D
Neuromuscular Junction Disorders Abdullah Al-Salti R3 AHD 9 march 2011
Myasthenia Gravis Myasthenia Gravis: a chronic autoimmune neuromuscular disease characterized by varying degrees of weakness of the skeletal (voluntary) muscles of the body. Age and Gender: • Myasthenia gravis presents at any age. Female incidence peaks in the third decade of life, whereas male incidence peaks in the sixth or seventh decade. • The female-to-male ratio is said classically to be 6:4, but as the population has aged, the incidence is now equal in males and female Inheritance: • 1st degree relatives have 1000x the rest of general population • inc. jitter on SFEMG demonstrated in 33-45% of asymptomatic 1st degree relatives • Inc. titres of AChR antibodies in up to 50%
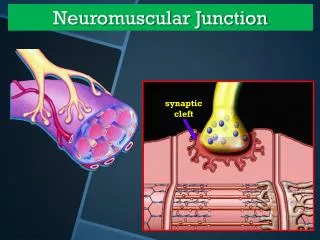
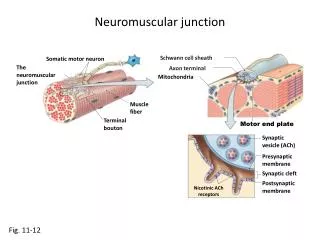
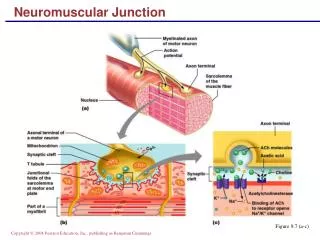
Pathophysiology Immunoglobulin G (IgG) directed attack on the NMJ, aimed specifically at the nicotinic acetylcholine (ACH) receptor. Damage to the ACH receptor and postsynaptic membrane involves several steps. • First, binding of the antibody to the receptor can directly block the binding of ACH. • Second, there is a complement-directed attack, with destruction of the ACH receptor and post junctional folds. • Third antibody binding can result in an increase in the normal removal of ACH receptors from the postsynaptic membrane. Resulting in a smaller endplate potential and a reduced safety factor of NMJ transmission.
Pathophysiology • The MuSK • maintains the normal functional integrity of the NMJ • anti-MuSK antibodies may alter the normal maintenance of a high density of AChRs at the NMJ • leading to reduced numbers of functional AChRs. • Thymus gland. greater than 50% of anti– AChR-positive patients having thymic hyperplasia and 10% to 15% having a thymic tumor.
HLA-DRW3, -B8, and -A1 predispose • a-subunit is the major T cell antigen
Etiology • Acquired autoimmune • Drug-induced : D-penicillamine • Transient neonatal (passive transfer of maternal anti-AChR antibodies) • Congenital myasthenic syndrome
Clinical presentation • most commonly presents with weakness of extraocular muscles ptosis and/or diplopia, +/- photophobia • can mimic any pattern of ophthalmoplegia including pupil sparing IIIrd nerve palsy, internuclear ophthalmoplegia (INO) or sixth nerve palsy • Bulbar involvement common eventually dysphagia, dysarthria, dysphonia (hypernasal or hoarse) • reduced facial expression, jaw fatigue • generally progresses over time so that within 2 years of onset of ocular MG, 90% have bulbar and proximal symmetric limb weakness
Clinical presentation Early Symptoms • frequent purchase of new eyeglasses to correct blurred vision • sleepy or sad facial appearance • avoidance of difficult to chew foods • cessation of activities requiring specific muscles (e.g. singing) Presenting Symptoms • ptosis or diplopia (2/3 of patients) • present in almost all within 2 years of onset • difficulty chewing, swallowing, talking (1/6) • limb weakness (1/10) • Rarely limited to single muscle group Diurnal Variation Exacerbating Factors
Clinical presentation Natural History • Restricted to ocular muscles in 10% of patients. • 90% have progressive weakness over two years involving oropharyngeal and limb muscles. • 2/3 of patients reach remission within 1 year • Spontaneous remission can occur, usually early Stages • Active stage • brief period of fluctuation • more severe • Inactive stage • fluctuations attributable to fatigue, intercurrent illness • Burnt out stage (15-20 years) • fixed weakness • atrophic muscles
Physical Findings Ocular Muscles • Ptosis • assymetrical • covering ptotic lid may relieve contraction of opposite frontalis • passively lifting ptotic lid may cause opposite lid to fall • varies during sustained activity • may shift from eye to eye - pathognomonic • edrophonium response EOMs • ³N muscle without pupil involvement • variable, fluctuating, and fatigable • most severe in medial rectus • “pseudo-INO” • edrophonium may improve only one of several weak ocular muscles
Physical Findings Oropharyngeal Muscles • altered facial appearance – depressed , snarl/sad • examiner can manually open jaw against resistance • impaired strength of eye closure • nasal regurgitation • difficulty swallowing • hoarseness (larynx) • nasal voice, especially after prolonged talking Limb Muscles • neck flexors > extensors • deltoids, triceps, wrist extensors, finger extensors especially affected • ankle dorsiflexors preferentially affected • fatigability
Tensilon test Measures • ptosis, limitation of eye movement, or nasal speech preferred • limb muscles (requires maximum effort) Administration • 2mg then wait 60s then 8mg then wait 60s • total dose 0.15mg/kg in children • sc administration in newborns and infants (delayed 2-5 minutes) Test Characteristics • 90% sensitivity not 100% specificity motor neuron disease oculomotor nerve palsies Alternative Agents • im neostigmine (longer duration of action) • therapeutic trial of pyridostigmine
Antibodies Against Acetylcholine Receptors Value confirm diagnosis but cant predict severity neither predict thymoma Test Characteristics • 80% sensitivity for generalized myasthenia (range 74-99%) • 55% sensitivity for ocular myasthenia • may rise with disease progression • generally specific • rare DDx SLE Inflammatory neuropathy (CIDP) ALS RA Thymoma without MG Normal relatives of patients with MG
Ocular cooling • Specificity?? • Positive 80%, no positive response in patients with ptosis not due to myasthenia • Alternative to tensilon test • Improvement in lid ptosis after the eye is cooled with ice pack 2 minutes
Repetitive nerve stimulation • (CMAP) should be normal in patients with MG. • With a small CMAP, LEMS should be considered. • Reproducible 10% decremental in amplitude when the first stimulus is compared to fourth or fifth • skin temperature (i.e. 32ºC – 34ºC) before beginning RNS. • hold anticholinesterases before test • more common in proximal muscles (facial, biceps, deltoid, trapezius) • may get some repair with exercise, but not an increment (unlike LEMS) • RNS in hand/shoulder has sensitivity of 61% • 76% in generalized myasthenia • 48% in ocular myasthenia
SFEMG (MUAP) abnormalities suggestive of a NMJ disorder,unstable MUAP, small, short-duration. • At least one symptomatic muscle • Increased jitter and blocking • Jitter is greatest in weak muscles • Measure 2 time locked motor units • SENSITIVE: 99% sensitive for generalized and 97% sensitive for ocular • NONSPECIFIC • any other motor unit disease • must perform EMG and NCS to exclude neuronopathy, neuropathy, myopathy • limb jitter DOES NOT predict development of generalized myasthenia • should examine 20 pairs in each muscle
Comparison of Diagnostic Techniques • Tensilon test diagnostic if positive in patients with ptosis or ophthalmoparesis • AChR Ab specific • RNS confirms DNMJ transmission but nonspecific; often normal in mild or ocular disease • SFEMG sensitive but not specific Other Diagnostic Procedures • CT chest • TFTs • TB test prior to immunosupression
Treatment Cholinesterase Inhibitors Roles • diagnostic • early, symptomatic treatment • adjunct to immunomodulatory and immunosuppressive therapy. • RARELY can be used as chronic treatment • usually effect diminishes with time Dosing Pyridostigmine • longer duration of action • initially 30-60mg q4-8 hours • 1.0mg/kg in infants and children • available in serum (60mg/5ml) • available as nebulizer • RARELY produces normal strength and RARELY completely corrects diplopia
Treatment Cholinesterase Inhibitors Neostigmine • initially 7.5-15.0mg q4-8h • 0.3mg/kg in infants and children • available as IV and nebulizer Side Effects Smooth Muscle (muscarinic) • NxVx • cramps • diarrhea • Rx with loperamide, propantheline, glycopyrrolate, diphenozylate Autonomic (muscarinic) bronchial/oral secretions
Treatment Cholinesterase Inhibitors Skeletal Muscle (nicotinic) • weakness Bromism (from Mestinon) • acute psychosis • rash • measure bromine level Drug Interactions succinylcholine metabolized by acetylcholinesterase impaired metabolism can lead to potential arrythmias
Corticosteroids Efficacy • marked improvement or complete relief in 75% • some improvement in 25% • most improvement in 6-8 weeks • may become total remission even later • best response if treated early • severity does not predict response • respond better with shorter disease duration and younger <50 Initiation - standard • begin with 1.5-2.0mg/kg/day • given until sustained improvement (2 weeks) then changed to 100-120mg alternate days Initiation - alternative • begin at 20mg qd • increase in 10mg increments q1-2 weeks • then maintain constant until improvement is maximum • used in ocular myasthenia
Corticosteroids Taper decrease to lowest dose necessary to maintain improvement • by 20mg each month until dose is 60mg • by 10mg each month until dose is 20mg • by 5mg each 3 months until 10 every other day • if weakness occurs, increase prednisone or add another immunosuppressant • do not d/c altogether Initial Worsening • 1/3 of patients • within 1st 7-10 days and lasts 6 days • cover with ChE inhibitors • use initial PLEX and do in hospital if oropharyngeal weakness or respiratory insufficiency
Immunosupressants Azathioprine Regimen • start 50mg/d • +50mg/d q7d • target 150-200mg/d Efficacy • improvement maintained • ?synergistic with steroids • may start simultaneous with steroids and taper steroids when azathioprine kicks in • effect takes 4-8 months • maximum improvement within 12 months • 70-90% response rate – similar to steroids
Immunosupressants Azathioprine • severe allergic reaction (required discontinuation) • 2 weeks after initiation • 15-30% • flu-like symptoms and rash • GI irritation (use divided doses after meals or decrease dose) • leucopenia (monitor CBC q1wk x1m, q1m x1y, q3-6m after) • Transaminitis • stop only if >2x normal and can restart at lower dose once normalize • no proven increase in malignancy in MG patient specifically (unlike in SLE)
Immunosupressants Cyclosporine Regimen • begin at 5-6mg/kd div q12h • trough levels after 1 month (allows tissue saturation) - aim for 75-150ng/ml Efficacy • improvement in most patients taking CYA • improve within 1-2 months • maximum improvement at >6months SFX • Renal toxicity . • HTN (monitor q1month until steady state) • ++drug interactions
Immunosupressants Cyclophosphamide Use • severe, refractory MG Regimen • 200mg/m2 q1month • titrate to changes in strength and side effects • 150-200mg/d po to total of 5-10g to relieve Sx SFX • alopecia • cystitis • leucopenia • NxVx anorexia
Immunosupressants Mycophenolate Mofetil Mechanism • Selectively inhibits proliferation of B- and T- lymphocyte clones responding to antigenic stimulation • Suppresses formation of antibodies Evidence • Open label pilot study demonstrated role as adjunctive therapy in refractory MG Dosing • 2g/d div bid Efficacy • Improvement as early as 2 weeks and usually seen within 2 months Role • Refractory MG • Steroid sparing agent when imuran intolerable or ineffective Side Effects • Diarrhea • leukopenia
Plasma Exchange Roles • sudden worsening of myasthenic symptoms for any reason • rapidly improve strength before surgery • concomitantly with high dose steroids • chronic intermittent Rx in refractory MG Protocol 2-3L of plasma 3x per week until improvement plateaus (usually after 5-6 exchanges) Adverse Effects • cardiac arrhythmias • Lightheadedness • Chills • Obscured vision • Pedal edema • Hemorrhage (removal of coagulation factors) • Hypercoagulation (removal of antithrombin III) • Coagulation defects corrected within24h
IVIG Indications • Similar to PLEX Mechanisms • blocks Fc receptors on macrophagesm • anti-idiotype Ab against AChR antibodies Protocol 2g/kg over 2-5 days SFX Headache , fever and chills, alopecia, aseptic meningitis, leucopenia retinal necrosis, renal failure.
Transitory Neonatal Myasthenia clinical features • hypotonic • onset within hours of birth but can be delayed up to 3 days • feed poorly in 1st 3 days • can get weak cry and lack of facial expression in 50% • 15% have limited EOM and ptosis • respiratory insufficiency rare • worsens for first few days then improves • last 2-12 weeks (usually 2 weeks) • neonatal antibodies have half life of 2-3 weeks and not detected after 5 months • recovery is complete
Transitory Neonatal Myasthenia diagnosis • tensilon test OR RNS • high AChR in neonate blood treatment • ChE for swallowing or breathing - just before feeding • neostigmine im before feeding • can also give via NG tube at 10 times parenteral level • PLEX if respiratory weakness (rare).
Anti-Musk Myasthenia Epidemiology • 25% of all MG patients are seronegative • 40% of seronegative MG patients are MuSK positive (about 10% overall) • M=F • Same age of onset as usual myasthenia Normal Function of MuSK • Tyrosine kinase • Regulates and maintains AChR at NMJ Clinical Features – Differences from Typical Myasthenia • More involvement of neck, shoulder, respiratory • Less limb • More bulbar • Increased risk of myasthenic crisis in 1st 2 years • Greater proportion in more severe category • Outcome similar, but require more steroids Treatment • Treatment generally the same • Can respond to thymectomy • Less likely to have thymic hyperplasia
Genetic Myasthenic SyndromesCongenital Myasthenia Genetics several genetic defects Epidemiology 2:1 male predominance Causes • Deficiency of muscle acetylcholine receptors at the end plate • Some have AChR mutations • Some have deficiency of rapsyn (receptor-associated protein at the synapse) • Abnormalities of acetylcholine resynthesis or immoblilization • Reduced end plate acetylcholinesterase • Impaired AChR function
Genetic Myasthenic SyndromesCongenital Myasthenia Clinical Features • ophthalmoparesis and ptosis developing in infancy • incomplete at onset • progresses to complete paralysis during infancy or childhood • mild facial paresis • limb weakness mild compared to opthalmoplegia • respiratory distress unusual • symptoms may not fluctuate much
Genetic Myasthenic SyndromesCongenital Myasthenia Diagnosis • subcutaneous injection of edrophonium transitory improvement in ocular motility • RNS • Decrement found in some limb muscles • May be necessary to test proximal or facial muscles if limbs normal • SFEMG Treatment • ChE inhibitors improve limb weakness in many forms • Ocular muscle weakness less responsive • Some children respond to DAP
Congenital Myasthenic Syndrome with Episodic Apnea (Familial Infantile Myasthenia) Clinical Features • Similar problems in other siblings • generalized hypotonic at birth +/- arthrogryposis • respiratory insufficiency and feeding difficulty at birth • repeated episodes of life-threatening apnea and feeding difficulty neonatally • may require ventilation • usually improves within weeks of birth, allowing weaning from ventilation • episodes may persist throughout infancy and even into adulthood • sudden bouts of respiratory distress with intercurrent illness • ocular function usually normal