Download

1 / 2
20 likes | 167 Views
x. Various scoring criteria were used to combine the two separate domains of the α7 nAChR, The ligand binding domains modelled on AChBP from Lymnaea stagnalis and the transmembrane domain is modelled on the TM of Torpedo marmorata (1OED).

E N D
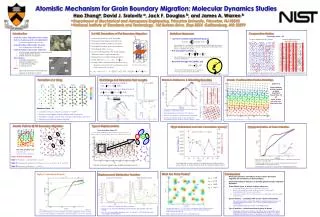
x Various scoring criteria were used to combine the two separate domains of the α7 nAChR, The ligand binding domains modelled on AChBP from Lymnaea stagnalis and the transmembrane domain is modelled on the TM of Torpedo marmorata (1OED) 1 332 664 996 1328 1660 Residue number 1 332 664 996 1328 1660 Residue number Possible gate region Ligand binding site Molecular Modelling Studies of the α7 nAChRBy: Shiva Amiri, Kaihsu Tai, Oliver Beckstein, Philip Biggin, Mark S.P. SansomDepartment of Biochemistry, University of Oxford, http://sansom.biop.ox.ac.uk contact: shiva.amiri@bioch.ox.ac.uk A – constructing the models • Results • Our alignment program allowed us to construct a complete model of the α7 nAChR from its separate domains. The ligand binding domain was modelled using the Lymnaea stagnalis protein AChBP (1I9B) and the transmembrane (TM) domain was modelled on the Torpedo marmorata TM domain (1OED). We analyzed the α7 nAChR model with coarse-grain methods (GNM and CONCOORD), electrostatic calculations, and pore profiling. • GNM identified the ligand binding regions as highly flexible. With CONCOORD we were able to perform principle component analysis (PCA) to look at the plausible dominant motions of the protein using eigenvectors. These eigenvectors suggest an overall twisting motion of the top and bottom of the α7 nAChR which may present during the gating of the receptor. • Introduction • Membrane proteins make up approximately 25% of the human genome and are targets for over 90% of drugs being produced. • Only, only a very small percentage of these proteins have their structures resolved due to difficulties in obtaining good crystals in the presence of a lipid bilayer. • Here we present computational methods for determining the structure of membrane proteins and using the generated models to study their structural variability and possible motions in order to gain understanding of their function. • The structure of the α7 nicotinic acetylcholine receptor (nAChR) is generated and studied through coarse-grain methods and molecular dynamics simulations. Covariance matrix constructed using CONCOORD’s data showing residues which move together (covaried). Red suggests high covariance and blue shows anti-covariance. There plot shows intra-subunit and inter-subunit motion of the subunits of the α7 nAChR B – analysis of models • Methods • Homology modelling is used to generate models of separate parts of proteins. • We have written a program which aligns the separate domains of the protein using several scoring criteria specific to the protein being studied. Pore profile of α7 nAChR using HOLE, the gate region is identified by having the smallest diameter. • We will be using these models to carry out MD simulations, currently we are performing MD on the separate domains of the α7 nAChR. The first eigenvector from PCA using CONCOORD’s output. suggests an overall twisting motion may be involved in the gating of the α7 nAChR. Coarse-grain analysis using GNM. The flexible regions identified by GNM are the ligand binding sites and the outer alpha helices of the TM domain. • The generated models are used in various coarse-grain and other molecular modelling studies. • Coarse-grain analysis includes: Gaussian Network Models (GNM) to look at the flexible regions of proteins. CONCOORD generates random structures within distance restraints to give possible motions of the generated structures. • Electrostatic calculations such as Born profiles can be carried out on the generated models. Pore profile analysis is done with the program HOLE. • Docking analysis is performed to study the interaction between ligands (nicotine and carbamylcholine) and the corresponding proteins using AUTODOCK. • Molecular dynamics simulations with GROMACS – analysis on the atomic level • Summary • We have developed a method to construct complete models of proteins where only partial structural information is available. • The generated models are used in several levels of analysis from coarse-grain methods where possible motions can be hypothesized to Molecular dynamics which looks at atomistic detail. • This method can be applied to other proteins and protein complexes (i.e. AcrB + TolC) and other ligand-gated ion channels have also been modelled (GABA, Glycine, and Serotonin receptors). Born energy of a monovalent cation (solid line) along the length of the α7 nAChR Molecular dynamics studies of the AChBP structure (ligand binding domain homologue). Strong correlation is seen in the domains where the ligand carbamylcholine was bound. Covaried sections move together and may contact the TM domain to result in the gating of the receptor. Acknowledgements: thanks to everyone in the Sansom group, especially Professor Mark Sansom and Dr. Philip Biggin. Also the Wellcome Trust and OSAP for funding.
homology modelling structure of separate domains alignment of separate domains parameter space to be scanned parameter scan bad contacts scoring to find optimal model termini distances other criteria … stereochemical check and energy minimization final model analyses