Download
1 / 47
500 likes | 702 Views
Carbon-Carbon Bond Formation and Synthesis. Organometallic Compounds. Recall: two extremely important reactions of metals and organometallic compounds:

E N D
Organometallic Compounds • Recall: two extremely important reactions of metals and organometallic compounds: • Oxidative addition: The addition of a reagent to a metal center causing it to add two substituents which extract two electrons from the metal and increasing its oxidation state by two. • Reductive elimination: The elimination of two substituents which donate two electrons to the metal center causing the oxidation state of the metal to decrease by two.
Heck Reaction Overall: A palladium-catalyzed reaction in which the R group of RX, a haloalkene or haloarene, is substituted for a vinylic H of an alkene.
Heck Reaction (consider the alkene) • Substitution (H R) is highly regioselective; most commonly at the less substituted carbon of the double bond. • Substitution is highly stereoselective; the E configuration is often formed almost exclusively. E Less substituted, H Ph substitution occurs here. Neither E nor Z
Heck Reaction (RX = Haloalkene) • For RX = haloalkene: Reaction is stereospecific; the configuration of the double bond in the haloalkene is preserved. E E
Heck Reaction. Some considerations. • The catalyst: • most commonly Pd(II) acetate. • reduced in situ to Pd(0). • reaction of Pd(0) with good ligands gives PdL2. • The organic halogen compound: aryl, heterocyclic, benzylic, and vinylic iodides, chlorides, bromides, and triflates (CF3SO2O-). • alkyl halides with an easily eliminated b hydrogen are rarely used because they undergo b-elimination to give alkenes. • OH groups and the C=O groups of aldehydes, ketones, and esters are unreactive under Heck conditions.
Heck Reaction. More… • The alkene • The less the crowding on the alkene, the more reactive it is. • The base • Triethylamine, sodium, and potassium acetate, and sodium hydrogen carbonate are most common • The solvent. • Polar aprotic solvents such as DMF, acetonitrile, and DMSO. • aqueous methanol may also be used. • The ligand • Triphenylphosphine, PPh3, is one of the most common.
Heck Reaction Start here L = PPh3 R 0 II II II II Rotation about the C-C bond. This is where the R is swapped in, replacing the H.
Heck Reaction • The usual pattern of acyclic compounds: replacement of a hydrogen of the double bond with the R group. • If the R group has no H for syn elimination, then a b H may be abstracted elsewhere. This b H should be brought into position for syn elimination with the Pd. Can’t happen due to cyclohexane ring.
Suzuki Coupling Suzuki coupling: A palladium-catalyzed reaction of an organoborane (R’-BY2) or organoborate (RB(OMe)2) with an alkenyl, aryl, or alkynylhalide, or triflate (R-X) to yield R-R’. Overall:
Suzuki Coupling • Recallboranes are easily prepared from alkenes or alkynes by hydroboration. • Borates are prepared from alkyl or aryl lithium compounds and trimethylborate. PhCl + Li
Suzuki Coupling • These examples illustrate the versatility of the reaction.
Suzuki Coupling Reductive elimination Oxid. Addn Transmetalation R1 and OtBu swap Substitution
Alkene Metathesis • Alkene metathesis: A reaction in which two alkenes interchange carbons on their double bonds. • If the reaction involves 2,2-disubstituted alkenes, ethylene is lost to give a single alkene product.
Alkene Metathesis • A useful variant of this reaction uses a starting material in which both alkenes are in the same molecule, and the product is a cycloalkene. • Catalysts for these reactions are a class of compounds called stable nucleophilic carbenes.
Stable Nucleophilic Carbenes • Carbenes and carbenoids provide the best route to three membered carbon rings. • Most carbenes are highly reactive electrophiles. • Carbenes with strongly electron-donating atoms, however, for example nitrogen atoms, are particularly stable. • Rather than being electron deficient, these carbenes are nucleophiles because of the strong electron donation by the nitrogens. • Because they donate electrons well, they are excellent ligands (resembling phosphines) for certain transition metals. • The next screen shows a stable nucleophilic carbene.
Nucleophilic Carbene • A stable nucleophilic carbene.
Alkene Metathesis Catalyst • A useful alkene methathesis catalyst consists of ruthenium, Ru, complexed with nucleophilic carbenes and another carbenoid ligand. • In this example, the other carbenoid ligand is a benzylidene group.
Ring-Closing Alkene Metathesis • Like the Heck reaction, alkene metathesis involves a catalytic cycle: • Addition of a metallocarbenoid to the alkene gives a four-membered ring. • Elimination of an alkene in the opposite direction gives a new alkene.
Ring-Closing Alkene Metathesis Initiation Step Cycle start
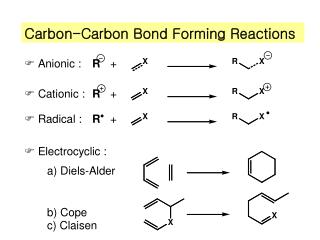
Diels-Alder Reaction • Diels-Alder reaction: A cycloaddition reaction of a conjugated diene and certain types of double and triple bonds. • dienophile: Diene-loving. • Diels-Alder adduct: The product of a Diels-Alder reaction.
Diels-Alder Reaction • Alkynes also function as dienophiles. • Cycloaddition reaction:A reaction in which two reactants add together in a single step to form a cyclic product.
Diels-Alder Reaction • We write a Diels-Alder reaction in the following way: • The special value of D-A reactions are that they: 1. form six-membered rings. 2. form two new C-C bonds at the same time. 3. are stereospecific and regioselective. Note the reaction of butadiene and ethylene gives only traces of cyclohexene.
Diels-Alder Reaction • The conformation of the diene must be s-cis.
Diels-Alder Reaction Steric Restrictions • (2Z,4Z)-2,4-Hexadiene is unreactive in Diels-Alder reactions because nonbonded interactions prevent it from assuming the planar s-cis conformation.
Diels-Alder Reaction • Reaction is facilitated by a combination of electron-withdrawing substituents on one reactant and electron-releasing substituents on the other.
Diels-Alder Reaction • The Diels-Alder reaction can be used to form bicyclic systems.
Diels-Alder Reaction • Exo and endo are relative to the double bond derived from the diene.
Diels-Alder Reaction • For a Diels-Alder reaction under kinetic control, endo orientation of the dienophile is favored.
Diels-Alder Reaction • The configuration of the dienophile is retained.
Diels-Alder Reaction • The configuration of the diene is retained.
Diels-Alder Reaction • Mechanism • No evidence for the participation of either radical of ionic intermediates. • Chemists propose that the Diels-Alder reaction is a concerted pericyclic reaction. • Pericyclic reaction: A reaction that takes place in a single step, without intermediates, and involves a cyclic redistribution of bonding electrons. • Concerted reaction: All bond making and bond breaking occurs simultaneously.
Diels-Alder Reaction • Mechanism of the Diels-Alder reaction
Aromatic Transition States • Hückel criteria for aromaticity: The presence of (4n + 2) pi electrons in a ring that is planar and fully conjugated. • Just as aromaticity imparts a special stability to certain types of molecules and ions, the presence of (4n + 2) electrons in a cyclic transition state imparts a special stability to certain types of transition states. • Reactions involving 2, 6, 10, 14.... electrons in a cyclic transition state have especially low activation energies and take place particularly readily.
Aromatic Transition States • Decarboxylation of -keto acids and -dicarboxylic acids. • Cope elimination of amine N-oxides.
Aromatic Transition States • the Diels-Alder reaction • pyrolysis of esters (Problem 22.42) • We now look at examples of two more reactions that proceed by aromatic transition states: • Claisen rearrangement. • Cope rearrangement.
Claisen Rearrangement • Claisen rearrangement: A thermal rearrangement of allyl phenyl ethers to 2-allylphenols.
Cope Rearrangement • Cope rearrangement: A thermal isomerization of 1,5-dienes.
Cope Rearrangement Example 24.8 Predict the product of these Cope rearrangements.
Synthesis of Single Enantiomers • We have stressed throughout the text that the synthesis of chiral products from achiral starting materials and under achiral reaction conditions of necessity gives enantiomers as a racemic mixture. • Nature achieves the synthesis of single enantiomers by using enzymes, which create a chiral environment in which reaction takes place. • Enzymes show high enantiomeric and diastereomeric selectivity with the result that enzyme-catalyzed reactions invariably give only one of all possible stereoisomers.
Synthesis of Single Enantiomers • How do chemists achieve the synthesis of single enantiomers? • The most common method is to produce a racemic mixture and then resolve it. How? • the different physical properties of diastereomeric salts. • the use of enzymes as resolving agents. • chromatographic on a chiral substrate.
Synthesis of Single Enantiomers • In a second strategy, asymmetric induction, the achiral starting material is placed in a chiral environment by reacting it with a chiral auxiliary. Later it will be removed. • E. J. Corey used this chiral auxiliary to direct an asymmetric Diels-Alder reaction. • 8-Phenylmenthol was prepared from naturally occurring enantiomerically pure menthol.
Synthesis of Single Enantiomers • The initial step in Corey’s prostaglandin synthesis was a Diels-Alder reaction. • By binding the achiral acrylate with enantiomerically pure 8-phenylmenthol, he thus placed the dienophile in a chiral environment. • The result is an enantioselective synthesis.
Synthesis of Single Enantiomers • A third strategy is to begin a synthesis with an enantiomerically pure starting material. • Gilbert Stork began his prostaglandin synthesis with the naturally occurring, enantiomerically pure D-erythrose. • This four-carbon building block has the R configuration at each stereocenter. • With these two stereocenters thus established, he then used well understood reactions to synthesize his target molecule in enantiomerically pure form.