Download
1 / 14
140 likes | 262 Views
Protein structure prediction. Anttu Kurttio Ville Pietiläinen. Introduction. Proteins are one of the most important parts in any biological systems. Understanding the folding of the amino-acid chain to produce functional proteins is essential for studying cellular systems.

E N D
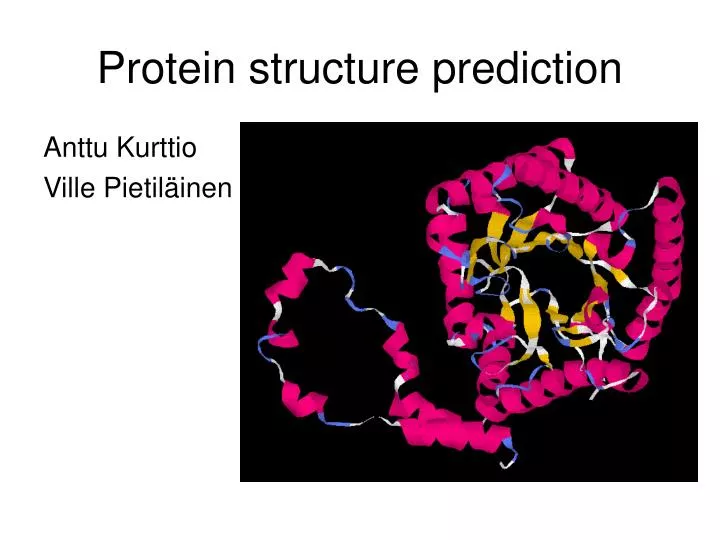
Protein structure prediction Anttu Kurttio Ville Pietiläinen
Introduction • Proteins are one of the most important parts in any biological systems. • Understanding the folding of the amino-acid chain to produce functional proteins is essential for studying cellular systems. • Fast and accessible methods of solving the 3D structure of a protein are in high demand.
Protein structure • This topic has been covered several times. Next!
Computational methods • Ab initio- methods • Laws of physics + amino-acid sequence = protein structure • Computes potential energy functions. • Minimum potential energy is the most stable structure and as such the most likely. • Computationally demanding.
Comparative methods • Based on the limited amount of possible tertiary structure types. • Approximately 2000 different types of protein folds. • Comparing the sample to a database of known structures, for example Protein Data Bank.
Homology modelling • Based on the assumption that homologous (related) proteins fold in a similar fashion. • Folding is a highly conserved factor, much more so than amino-acid sequence. • Finding a match between two distantly related proteins can be difficult.
Protein threading • Based on the assumption that similar folding has already been found. • Comparing parts of the sequence to a database of known three dimensional structures using a scoring function. • Works at least somewhat on approximately 80% of new protein sequences.
Programs • A lot of free programs are available. • Server based programs do the computational work. For example Swiss-Model, Rosetta or PSIPRED. • Downloadable applications are used for viewing the results. For example Swiss-PdbViewer or Rasmol. • Distributed computing promises increases in computational capacity.
DEMO • Swiss-Model in four easy steps. swissmodel.expasy.org
Rejoice! • Study your brand new model of a protein.