Download
1 / 11
150 likes | 277 Views
Multiple sequence alignment (msa). Motivation. “Two swallows do not make a summer” Discover conserved regions Predict important regions of the protein Discover domains Search for additional members of a protein family (profile-based searching) Build phylogenetic trees. Topics.

E N D
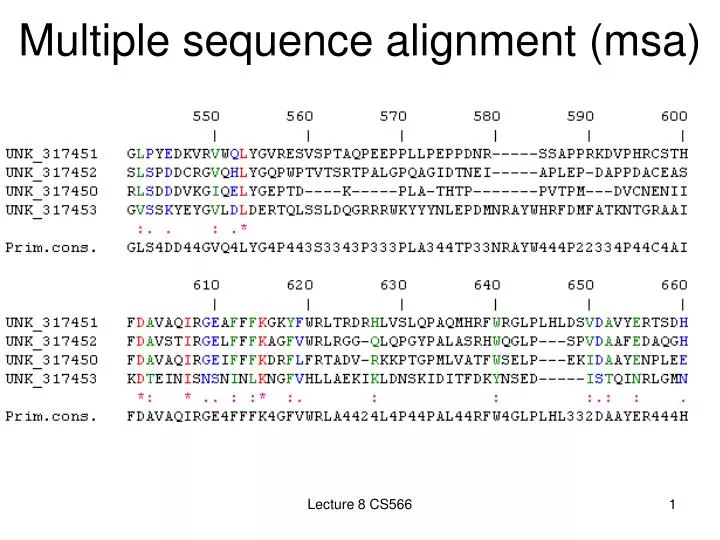
Multiple sequence alignment (msa) Lecture 8 CS566
Motivation • “Two swallows do not make a summer” • Discover conserved regions • Predict important regions of the protein • Discover domains • Search for additional members of a protein family (profile-based searching) • Build phylogenetic trees Lecture 8 CS566
Topics • Scoring schemes • Pairwise • N-way • Optimal • Multidimensional dynamic programming • Heuristic algorithms • Progressive • Iterative Lecture 8 CS566
Scoring schemes • Alignment score = lCl • Column Score Cl • Ideally • Based on n-way joint probability (n-generalized AAS) • Sum of Pairs • i<j sij Based on amino acid substitution matrices • Gap-gap = 0; Gap-char = -g • Commonest scheme used • Fallacious: • Assumes only 2-way and not n-way joint probabilities • Score not proportional to number of sequences in alignment • N-way sums • Need to know central point of reference (ancestral sequence) Lecture 8 CS566
Multidimensional Dynamic Programming • Line up n sequences in a grid having n dimensions • Score each cell as the maximum of • Lining up all corresponding characters AND • All possible combinations of gaps and characters • Note choice made • Reconstruct alignment by traceback • Global or Local dynamic programming? • Space complexity? • Time complexity? Lecture 8 CS566
MSA – Efficient Multidimensional Dynamic Programming • Carillo-Lipman MSA algorithm • Uses pair-wise dynamic programming to identify sub-matrix regions of near-optimality • n-dimensional dynamic programming carried out within space of intersection of near-optimal regions • Still limited to only a few sequences • Is this an optimal algorithm or not? Lecture 8 CS566
Progressive alignment • New concepts • Consider aligning alignments to alignments/sequences en bloc • Hierarchical/Sequentialorder of alignment (“Once a cobbler, always a cobbler”) • Heuristic • Fast Lecture 8 CS566
Progressive alignment - Clustal • Compute all pairwise alignments • Convert alignment scores into distances • Build guide tree (phylogenetic tree) • Align sequences in order suggested by ‘guide tree’ • Position specific scoring system used • Gap costs depend on position • Composition based scoring system used • Percentage similarity dictates choice of scoring matrix • Weighting based on composition bias • Only ‘cross-terms’ (profile-profile) used in scoring Lecture 8 CS566
Progressive alignment - Clustal • ClustalV (Now history!) • ClustalW (Takes weighting into account for composition bias) • ClustalX (Graphical interface) Lecture 8 CS566
Iterative refinement-1 • “Once a cobbler, now a king!” • Iterative algorithm: • Compute all pairwise similarities • Start with best pair • Add ‘most-similar’ sequence to profile successively till none left • Remove and re-align each sequence till convergence Lecture 8 CS566
Iterative refinement-2 • Genetic programming-based msa • Create initial random alignment • Score alignment • Retain better scoring half of alignment • Mutate remaining half of alignment with ideas from genetic recombination • Random gap insertion • En bloc shifts • Probabilistic order of alignment • Score resulting alignment • Iterate till convergence Lecture 8 CS566