Download

1 / 68
680 likes | 783 Views
"Everything You Always Wanted to Know about Computational Chemistry, But Were Afraid Would Be Answered by 27 Pages of Integrals in a Nomenclature That You've Never Seen Before." or

E N D
"Everything You Always Wanted to Know about Computational Chemistry, But Were Afraid Would Be Answered by 27 Pages of Integrals in a Nomenclature That You've Never Seen Before." or “How to Understand MO Calculations, for the Theoretically-Challenged." web.utk.edu/~bartmess/comptalk.html John Bartmess Dept. of Chemistry University of Tennessee
Theoretical Chemistry Molecular Orbital (MO) Calculations Quantum Mechanics Calculations Computational Chemistry "In theory, there is no difference between theory and practice. In practice, there is." - Jan L. A. van de Snepscheut (computer scientist) But often attributed to Yogi Berra "Man's gotta know his limitations" - Dirty Harry Callahan (John Milius)
Goal of this Talk: • To give you an understanding of the basics of • computational work in the literature • Information on which methods are really good, and • which are either inappropriate or flat-out garbage • for a given problem • - The Alphabet Soup of Computation
Goals of Computational Chemistry: Gas-phase molecules (molecules in solution take extra work, and involve major approximations) Geometries: Closed shell (= octets around all heavy (non-H) atoms): well-met even at low level calculations Open shell (= radicals, sextet cations): problematic, but solvable with knowledge Energies: relative versus absolute accuracy and precision
Other Quantities: Dipole Moments Orbital energies and occupations (eigenvalues and eigenvectors) Charge distributions (atomic and orbital) Mulliken populations (atomic charges) Spin matrices, total spin states Bond orders Ionization energy & electron affinity (vertical and adiabatic): Koopmans Physica19341, 104) Polarizabilities, hyperpolarizability Vibrational frequencies/force constants (intensities) Rotational constants/moments of inertia Entropy, Heat capacity, Partition functions Zero point energies
Units Bohr - One atomic unit of distance = 0.5292 Angstrom (archaic now) Hartree - One atomic unit of energy = 2 x IE(H.) 2625.500 kJ/mol 627.5095 kcal/mol 27.2114 eV219474.6 cm-1 Energetic Data (ab initio) - absolute energy, as negative value: cleavage of all bonds to form free atoms, then ionization of atoms to bare ionic nuclei plus free electrons at infinite distance (E = 0) benzene: -231.820 hartrees = -145,469 kcal/mol - atomization energy to atoms: benzene -2.1099 hartrees (-1324 kcal/mol); expt: -1323 kcal/mol - heat of formation semi-empirical: close (± 2 kcal/mol, average organics) ab initio: usually too unstable, unless very high level calculation (variational principle)
Practicalities speed ("cost" to computationalists): scales as as a high power of the number of electrons (typically n4 to n8) known failure modes of method (certain structures known to be wrong energy or geometry) cost of hardware 3.3GHz duo hex-core processor PC, 12 GB RAM, 1 TB hard drive : $3000 (Feb 2012) : 100x speed of a 1969 Cray I ($30M in current $) : 330,000x speed of Osborne (1979; $6K current) 10,000,000x media storage 200,000,000x RAM = 3 x1020 better, at ½ cost cost of software Gaussian 03 $1500 (site license) MOPAC (QCPE $400?) MNDO: free Linux (Red Hat or Fedora) for ab initio
Hierarchy of 4 Methods - Molecular Mechanics: Not a quantum mechanical method. - Empirical: Hückel, Extended Hückel - Semi-empirical archaic: INDO, PPP, CNDO/n, MINDO/n current: MNDO, AM1, PM3 - ab initio e.g. Gaussian, GAMES, MOLPRO (programs) Hartree/Fock Electron Correlation Configuration Interaction Extrapolation Density Functional Theory
Input name, method, time limits charge, multiplicity geometry: - Cartesian coordinates - Z matrix, or internal coordinates: bond lengths planar angles dihedral (torsional) angles connectivity sometimes called “Natural Coordinates”
Semi-empirical input AM1 precise acetone O C 1.22 1 1 C 1.54 1 120. 1 2 1 C 1.53 1 120. 1 180. 1 2 1 3 H 1.11 1 110. 1 180. 1 4 2 1 H 1.11 1 110. 1 60. 1 4 2 1 H 1.11 1 110. 1 -60. 1 4 2 1 H 1.11 1 110. 1 180. 1 3 2 1 H 1.11 1 110. 1 60. 1 3 2 1 H 1.11 1 110. 1 -60. 1 3 2 1 0 0 0 0 0 0 0 0 0 0 0
%mem=256MB %nosave # g3mp2b3 Opt=Maxcyc=100 Me2C(.)CH2NH3+ +1 2 N C 1 1.5283 C 2 1.5050 1 115.9692 C 3 1.5022 2 115.2106 1 182.2808 C 3 1.4956 2 124.5590 1 2.8544 H 4 1.1100 3 110.6428 2 60.1046 H 4 1.1100 3 110.6213 2 -239.9345 H 4 1.1077 3 112.2541 2 180.0153 H 5 1.1104 3 111.0735 2 61.1423 H 5 1.1108 3 111.3159 2 -239.8219 H 5 1.1085 3 111.9526 2 180.4718 H 2 1.1190 1 105.8385 3 122.4183 H 2 1.1192 1 105.6916 3 -122.5352 H 1 1.0250 2 110.0652 3 179.7632 H 1 1.0245 2 112.2413 14 119.2315 H 1 1.0246 2 112.1792 14 -119.2190
Common to all: - Input of starting geometry - Trial orbital set (Extended Hückel) - Self Consistent Field (modify orbitals to reflect reality) - Geometry Optimization (modify nuclear geometry to find minimum energy) Method of Steepest Descent – derivatives of E vs. geom. Problems Local minima: benzene with 1 H inside = +156 kcal/mol above reality Oscillation - Final output: Total energy, other properties Vibrational Frequencies, other Statistical Mechanics properties
Global vs. local minima: anti vs gauche butane Benzene, with H in center: ΔfH = 131 vs. 19.4 kcal/mol normal
Negative freq ↓ ←1/2hυ
Thermochemistry (non-0K) From statistical mechanics: Etot = E0 + Etrans + Erot + Evib + Eelec mass geometry (moments of inertia) vibrational frequencies orbital energies allow calculation of: zero point energy = h/2· E0 = E0 + ZPE heat capacity: E298 H298 (=E298 + RT) entropies: S298 G298
Frequencies - Scaling: 0.896 HF/6-31G* 0.96 B3LYP - Harmonic approximation (parabola), yet real ones anharmonic • Lowest ones (<300 cm-1) most important to • stat. mech. entropy, yet worst known • Internal rotors, free vs. hindered • Ring breathing modes
Heats of Formation Absolute: ΔfHo(molecule) = E0 (molecule) - E0(atoms) + ΔfHoexptl(atoms) Relative: A + B = C + D EA EB EC ED ΔfH(A) = EA+ EB - EC – ED + ΔfH(C) + ΔfH(D) - ΔfH(B)
EXACT THEORY: the Schrödinger Equation H(Ψ) = E·Ψwhere Ψ is a "full molecule" wave function.H = Hamiltonian function (general case: Hermetian operator) E = eigenvalue H = T (kinetic part) + V (potential part) M M M H = - h2/82 MA-12A + e2ZAZBrAB-1 A=1 A=1 B>A NN M N N - h2/(8m) i2 - 2ZArAi-1 + e2rij-1 i=1i=1 A=1 i=1j>i
Hamiltonian divides up into: • 1. Kinetic energy of nuclei • 2. Nuclear-nuclear repulsion • 3. Kinetic energy of electrons • 4. Nuclear-electron attraction • 5. Electron-electron repulsion • Born-Oppenheimer approximation: • Nuclei don’t move, on electron motion timeframe • 1. = 0 • 2. Static calculation: Coulomb’s Law
MORE APPROXIMATIONS: 1. Ψ = ψ1 . ψ2 . ψ3 ...., where ψi are one electron molecular orbitals. Separate the Schrödinger equation: H(ψi) = Ei· ψi 2 = probability of electron position All physical observables relate to 2, because has imaginary parts. Normalized: aa* = 1 <a|a*> = 1 Orthogonal: ab = 0 <a|b> = 0 no overlap
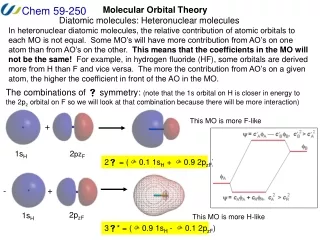
1 electron orbitals so far • Spin: and • = spatial· Pauli Principle: is antisymmetric wrt exchange of 2 electrons (1,2) = -(2,1) If every electron has its own , “unrestricted” If paired s, “restricted” (faster calculation)
2. Represent each ψi as a Linear Combination of Atomic Orbitals (LCAO): ψi = ci,1·φ1 + ci,2·φ2 + ..... where φj are basis orbitals (usually atomic)
3. Variational Principle: For any approximate (one e-) ψi, Ei from the Schrödinger Equationis greater than the true Ei for the exact ψi. Thus ψi and cij are varied so as to minimize Ei, or δEi/δci,j = 0. The true value of the variational principle is that one knows when the calculation is getting closer to reality, because the energy is going down. There are other methods, such as Density Functional Theory, or certain types of electron Correlation, that are not variational.
4. Self Consistent Field (SCF) approximation. “Three Body Problem” ψi is calculated for one given electron interacting with the field of the nuclei plus an average smeared-out charge distribution of all other electrons. This ψi is then used as part of the average distribution as the next electron's ψi is found, and so on. After successive iterations result in an energy change of less than a given amount (ca. 1 cal), the Self Consistent Field is said to have converged, and that set of ψis is used as a valid wave function.
5. Hartree-Fock Limit. - Approximations 2 and 4 (LCAO and SCF) lead to Eo always too high. - If a small number of terms [limited number of basis orbitals] is used in (2), then the ψi will not be as good as with a larger number of terms. - As a sufficiently large number of terms (j>20, typically) is used, E approaches the "Hartree-Fock limit".
This Hartree-Fock limit still is only 90-95% of the way to the true energy, since the SCF approximation ignores : (1) "electron correlation", or the fact that the other electrons are not a statistical average, but moving, when calculating the SCF. (2) "configuration interaction" or "CI", because empty orbitals mix into filled MOs. (3) relativistic speed of the core electrons, which can still contribute a 0.1% error in total energy (especially important for atoms low in the Periodic Table)
RHF (Restricted Hartree-Fock) Every spatial orbital has an exactly equal orbital, i.e. every spin up electron has a spatially equivalent spin down electron. This generally implies a closed-shell wavefunction, though restricted open-shell SCF can be done. UHF (Unrestricted Hartree-Fock) Every spin-orbital has different spatial forms. Drawback: time, spin contamination. spin-contamination: calculations with UHF wavefunctions that are not eigenfunctions of spin, and are contaminated by states of higher spin multiplicity (which usually raises the energy).
ECP = Effective Core Potential. The core electrons have been replaced by an effective potential. Saves computational expense. May sacrifice some accuracy, but can include some relativistic effects for heavy elements. isodesmic: a chemical reaction that conserves types of chemical bond. MeO- + EtOH → MeOH + EtO- isogyric: a chemical reaction that conserves net spin. Lower-level calculations of such relative energetics can be as accurate as much higher(slower) ones of absolute energetics
Koopman's Theorem: IE = energy of the HOMO (Highest Occupied Molecular Orbital). This is a vertical IE, not adiabatic. Errors from no e- correlation plus geometry relaxation tend to cancel for IEs. EA = energy of the LUMO (Lowest Unoccupied Molecular Orbital). These errors compound for trying to approximate EA ______________ ______________ ------------------------ 0 E ______________ LUMO _____↑↓______ HOMO _____↑↓ _____
MERP (Minimum Energy Reaction Path) or IRC (Intrinsic Reaction Coordinate): An optimized reaction path that is followed downhill, starting from a transition state, to approximate the course (mechanism) of an elementary reaction step. (Ignores tunneling, contribution of vibrationally excited modes/partition function, etc.) Transition States: saddle points (one negative frequency), sometimes found as minima. Search routines exist. scaling: Multiplying calculated results by an empirical fudge factor in the hope of getting a more accurate prediction. Very often done for vibrational frequencies computed at the HF/6-31G* level, for which the accepted scaling factor is 0.893.
Molecular Mechanics Methods "Balls and Springs" MM2 - Allinger Force Field version 2 MM3 - MMX - PCModel Sybyl - Amber - CHARMn - All ΔfH ca.±1 kcal/mol μD±0.1 Limit: only parameterized functional groups Advantage: fast, up to proteins
Empirical Methods Hückel Calculation Many integrals pre-calculated or equated to measured data Pros: orbital symmetry resonance energy back of envelope Cons: flat geometry, π orbitals only polar bonds poor
EHT - Extended Hückel Theory (Roald Hoffman) Hückel with sigma bonds as well Ignores e- e- repulsion Uses expt’l IEs for certain integrals Pros: Ethane rotational barrier Woodward-Hoffman rules includes AO overlap terms Frontier orbitals All elements Cons: valence only (not hypervalents) geometry poor (Me-Me = 1.92Å) partial charges high singlet & triplet same (no e- spin) Used as first guess for higher level methods
Semi-Empirical Methods Approximation: many computationally expensive (= slow) integrals replaced by adjustable parameters, determined by fitting experimental atomic and molecular data. Non-nearest-neighbor interactions neglected Different choices of parameterization lead to different specific theories (e.g., MNDO, AM1, PM3). Archaic: CNDO - Complete Neglect of Differential Overlap PPP - Pariser-Parr-Pople INDO/1 - Intermediate NDO MINDO/3 – Modified Intermediate Neglect..
MNDO: Minimal Neglect of Differential Overlap Atoms: H, Li-F, Al-Cl, Cr, Zn, Ge, Br, Sn, I, Hg, Pb Basis: 32 molecule parameterization Developed by M.J.S. Dewar Problems (geometries): -O-O- bond ~0.17Å short C-O-C angle 9o large amides pyramidal Aniline, nitrobenzene: NH2, NO2 group perpendicular to ring, due to nuclear repulsion
MNDO Problems (energies): no H-bonds, no H2O dimer S, Cl, & Br Ionization Energies high activation barriers high bond dissociation enthalpies too weak conjugation too stable 3-center B bonds too stable no Van der Waals attraction: Sterically crowded hydrocarbons too unstable (Me4C: -24. kcal/mol, exp -40.3 kcal/mol) N-O bonds poorly parameterized - heats way off (MeNO2: calc ΔfH = +5.1, exp -17.9 kcal/mol) 4 membered rings too stable (cyclobutane: -11.9, exp +6.8 kcal/mol) (cubane: + 108 , exp 148.7 kcal/mol) Underestimates polarizability interactions (aliphatic alcohol acidities all the same) hypervalent unstable 3rd,4th row elements: only low valent cases have good absolute heats though relative heats of same oxidation state okay
AM1 - Austin Model 1 (Dewar) Atoms: H, Li, B - F, Al - Cl, Zn, Ge, Br, I, Hg Basis: 100 molecule parameterization Pros: H-bond energies, lengths better proton affinities good better activation barriers Heat of Formation 40% better 2-Cl-THP axial (anomeric effect) Aniline, nitrobenzene now planar
AM1:Problems: poor on hypervalent compounds (none in parameterization set) conjugate interactions low -CH2- ΔfH ~ 0.2 kcal/mole low each Heat of Hydrogenation low bond dissociation enthalpies too weak activation enthalpies high -NO2 energies high -O-O- bond ~ 0.17Å short H-bond angles, H2O H-bond geometry wrong C-C-O-H gauche in ethanol proton transfer barrier high
PM3 – Parameterized Model 3 (Stewart: student of Dewar’s) Program: MOPAC Atoms: H, Li, Be, C-F, Mg-Cl, Zn-Br, Cd-I, Hg-Bi Basis: 657 molecule parameterization Pros: hypervalent included in parameterization set ΔfH 40% better -NO2 better ground state geometries better H2O H-bonds: lengths & angles
PM3: Cons: partial charges on N unreliable bond dissociation enthalpies low amides pyramidal, barrier low no barrier to formamide rotation spurious minima D2d symmetry for CBr4 IEs poor proton transfer barrier high wrong glucose geometry: H-bonds 0.1A short C-C-O-H gauche in ethanol Van der Waals attraction high/H-H core repulsion low (MeNO2: calc -15.9, exp -17.9 kcal/mol) (cyclobutane: -3.8, exp +6.8 kcal/mol) (cubane: 114, exp 148.7 kcal/mol) (Me4C: -35.8, exp -40.3 kcal/mol) (MeOH..-OMe: bond strength 19, exp 28.8 kcal/mol Hypervalents good energy
Ab initio Methods Hartree-Fock methods Basis Set: math functions that describle orbitals STO (Slater-Type Orbital) Minimal Basis Set Basis function with an exponential radial function, i.e., e –αr or a fit to such a function using other functions, such as Gaussians: e-ar2 (Gaussians are computationally faster) STO-3G “stodgy” (1969, Pople) is a MBS that uses 3 Gaussians to fit an exponential. Exponentials are better basis functions than Gaussians, but are expensive computationally.
Split Valence: a basis set that is more than minimal for the valence orbitals. Much better for polar bonds than MBS. DZ (Double-Zeta): A basis set for which there are twice as many basis functions as are minimally necessary. "Zeta" (Greek letter ζ) is the usual name for the exponent that characterizes a Gaussian function. (Dunning, 1970) TZ: (triple zeta)
3-21G Basis set: 3 Gaussian function primitives for core electrons Split Valence: 2 Gaussians with linked coefficients for inner valence electrons 1 Gaussian for each outer valence electron - Polar bonds better described than minimal basis set - Atoms: H – Xe 6-31G Basis set: 6 Gaussian functions for core 3 Gaussian (linked coefficients) for inner valence electrons 1 Gaussian for each outer - Atoms: H - Ar
6-31G* = 6-31G(d) 6-31G plus a set of polarizing d-functions (6D) added to heavy atoms - most popular, widely used/validated - Atoms: H - Ar - Polarization functions help to account for the fact that atoms within molecules are not spherical. Even better for polar bonds. 6-31+G diffuse (large) s orbitals added (in essence opposite of *) - negative ions bound - slower 6-31+G* = 6-31+G(d) - Augmented 6-31G* 6-31++G* = 6-31++G(d) - Augmented 6-31+G set of diffuse s-functions added to H, too 6-31+G* = 6-31+G(d,p)- 6-31++G* = 6-31++G(d,p)-
cc-pVDZ - Correlation Consistent, polarized Valence Double Zeta Basis: correlation consistent basis set Valence Double Zeta set of polarizing d-functions (5D) added to heavy atoms Pros: use with correlated methods series converges exponentially to complete basis set limit Atoms: H-Ne, B-Ne, Al-Ar cc-pVDZ+ - Augmented cc-pVDZ Basis: add diffuse functions Atoms: H, C-F, Si-Cl cc-pVDZ++ cc-pVTZ - Correlation Consistent Valence, polarized Triple Zeta
Post-Hartree-Fock Methods • Electron Correlation: • Explicitly considering the effect of the interactions of specific electron pairs, rather than the effect each electron feels from the average of all the other electrons. (the latter is the SCF approximation). • Large correlation effects occur for: • electron rich systems • transition states • "unusual” coordination numbers • no unique Lewis structure • conjugated multiple bonds • radicals and biradicals
MP2 - 2nd Order Møller Plesset ( = Many Body Perturbation Theory) Basis: Taylor Series expansion, truncated at 2nd order Pros: dynamic correlation for Van der Waals forces: CH4 - CH4 binding π-π stacking interaction bond breaking consistent with diradical formation (without correlation, heterolytic cleavage is seen) anomeric effect Cons: not variational (MP3, MP4, etc.) transition metals not parametrized overbinds CO2, PO free radicals too stable O3 frequencies way off bonds too long scales as n5 (slow)