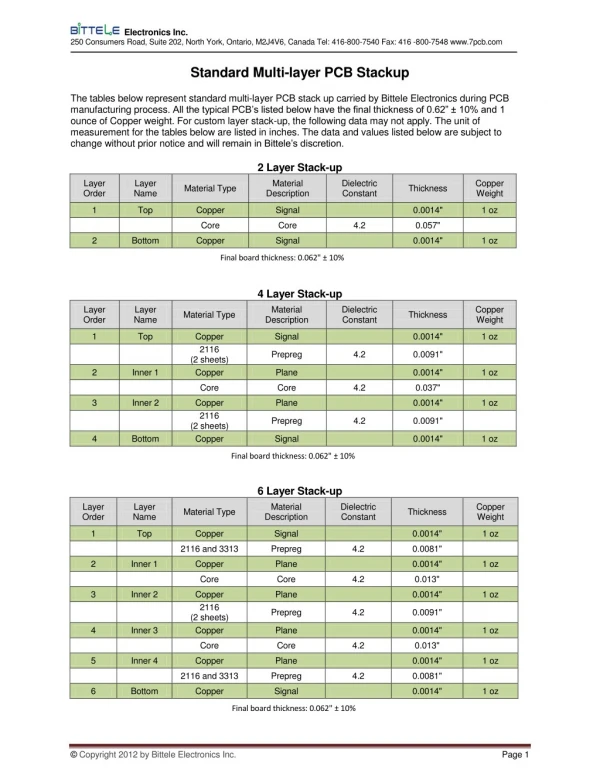
Download

1 / 20
200 likes | 377 Views
Comparison of multi-standard and TMS-standard calculated NMR shifts for coniferyl alcohol. Heath D. Watts, Mohamed N.A. Mohamed, James D. Kubicki 7 April 2011. Goal – Build a reasonably accurate model of lignin testable against spectroscopic data. www.lbl.gov/Publications/YOS/Feb/.

E N D
Comparison of multi-standard and TMS-standard calculated NMR shifts for coniferyl alcohol Heath D. Watts, Mohamed N.A. Mohamed, James D. Kubicki 7 April 2011
Goal – Build a reasonably accurate model of lignin testable against spectroscopic data www.lbl.gov/Publications/YOS/Feb/
Experimental 13-C NMR data for coniferyl alcohol in acetone Monomer provides less convoluted spectrum, but has ambiguous shifts g b a 1 2 6 Me 3 5 4 http://ars.usda.gov/Services/docs.htm?docid=10491
Can computational chemistry methods reproduce the observed NMR chemical shifts for coniferyl alcohol?
Energy minimization method: Structure B3LYP/6-311++G(d,p) Cheesemanet al. Journal of Chemical Physics. 1996, 104(14), 5497. NMR Theory: Chemical shielding B3LYP/6-311+G(2d,p); NMR standard: TMS Inorganic character d13C = sTMS - ssample Si
1:1 line MG5 (Watts, 2011) http://ars.usda.gov/Services/docs.htm?docid=10491
g b a 1 6 2 MG1 MG2 MG3 5 3 4 Me MG4 MG5 MG6
NMR Theory: mPW1PW91/6-31G(d); NMR standard: benzene sp2 C; CH3OH sp3 C Organic standards Multi-standard d13C = sM-S – ssample + exp,ref • Sarotti & Pellegrinet; Journal of Organic Chemistry. 2009, 74, 7254. NMR Theory: B3LYP/6-311+G(2d,p); NMR standard: TMS NMR Theory: HF/6-311+G(2d,p); NMR standard: TMS TMS, single standard d13C = sTMS - ssample Cheesemanet al. Journal of Chemical Physics. 1996, 104(14), 5497.
MG3 mPW1PW91/6-31G(d) Slope: 1.00 y-intercept (ppm): -0.42 r2=0.994 MUE (ppm): 2.2 RMSE (ppm): 2.4 ppm Max Error (ppm): 3.7
NMR Theory: mPW1PW91/6-31G(d); NMR standard: benzene sp2 C; CH3OH sp3 C NMR Theory: B3LYP/6-311+G(2d,p); NMR standard: TMS NMR Theory: HF/6-311+G(2d,p); NMR standard: TMS
Reviewer comments: …the authors conclude that the MG3 should be the “experimentally observable conformer”. In the case of flexible compounds, the generally accepted protocol is to calculate the Boltzmann-averaged shielding constants, which gives a more “realistic” result, because it takes into account the effect of all significantly populated conformations. In addition, the authors did not mention the relative energies of the different conformers.
The Gibbs free energy of solution (G°soln) was calculated by: • G°soln = G°IEFPCM + G°TCDG • G°IEFPCM total free energy in solution with all non-electrostatic terms from the polarized continuum calculation (solvents were acetone, DMSO, & CHCl3) • G°TCDG thermal correction to Gibbs free energy from the gas-phase frequency calculation • The relative G°soln for each model was determined by setting the model with the lowest G°soln to 0 kJ/mol (Foresman, 1996; www.gaussian.com/g_whitepap/thermo.htm)
Boltzmann-weighted NMR chemical shifts to account for contribution of each conformer based the energy distribution (Barone, 2002) • 13CX Boltzmann averaged chemical shift of atom X • 13CXi Chemical shift of atom X in conformer i Probability
MG3 only mPW1PW91/6-31G(d) Slope: 1.00 y-intercept (ppm): -0.42 r2=0.994 MUE (ppm): 2.2 RMSE (ppm): 2.4 ppm Max Error (ppm): 3.7
Conclusion: coniferyl alcohol • For d13C NMR calculations on coniferyl alcohol • Performance of multi-standard method >> TMS-standard method • Linear correlation • Statistical errors • Multiple, Boltzmann-weighted conformers better predict chemical shifts than did comparison of a particular conformer with data
Acknowledgments • USDA National Needs Graduate Fellowship Competitive Grant 2007-38420-17782 from the National Institute of Food and Agriculture to H.D. Watts through Nicole Brown. • Instrumentation funded by the National Science Foundation through grant OCI-0821527. • JDK, MNAM, and HDW acknowledge support of the U.S. Department of Energy grant for the Energy Frontier Research Center in Lignocellulose Structure and Formation (CLSF) from the Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001090. • HDW acknowledges support from Shell Geosciences Energy Research Facilities Award • MNAM was supported by the USDA grant “Improved Sustainable Cellulosic Materials Assembled Using Engineered Molecular Linkers” through Jeff Catchmark. • Computational support was provided by the Research Computing & Cyberinfrastucture group at the Pennsylvania State University. • Discussions with Ming Tien, Brett Diehl, Nicole Brown and other members of the Center for Nanocellulosics and CLSF are also acknowledged.
References • Adamo, C.; Barone, V. Journal of Chemical Physics. 1998, 108(2), 664-675. • Bachrach, S.M. Quantum mechanics for organic chemistry. In Computational Organic Chemistry. Wiley-Interscience: New Jersey, 2007, pp 1-42. • Barone, G.; Duca, D.; Silvestri, A.; Gomez-Paloma, L.; Riccio, R.; Bifulco, G. Chemistry—a European Journal. 2002, 8(14), 3240-3245. • Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993a, 98, 1372. • Becke, A. D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993b, 98(7), 5648. • Cheeseman, J.R.; Trucks, G.W.; Keith, T.A.; Frisch, M.J. Journal of Chemical Physics. 1996, 104(14), 5497-5509. • Foresman, J.B.; and Frisch, Frisch, Æ. Exploring Chemistry with Electronic Structure Methods. 2nd Ed. Gaussian, Inc.: Pittsburgh, PA, 1996. • Gill, P.M.W. Density functional theory (DFT), Hartree-Fock (HF), and the self-consistent field. In Encyclopedia of Computational Chemistry. von Ragué Schleyer, P.; Allinger, N.L.; Clark, T.; Gastiger, J.; Kollman, P.A.; Schaefer III, H.F.; and Schreiner, P.R., Eds. Wiley: New York., 1998, Volume 1, pp 678-689. • Grunenber, J., Editor. Computational Spectroscopy. Wiley-VCH: Weinheim, 2010. • Holtman, K.M.; Chang, H.; Jameel, H. J Wood Chem Tech. 2006, 26, 21-34 • Koch, W.; Holthausen, M.C. A Chemist’s Guide to Density Functional Theory. Wiley-VCH: New York, 2002. • Kubicki, J.D. Transition State Theory and Molecular Orbital Calculations Applied to Rates and Reaction Mechanisms in Geochemical Kinetics. In Kinetics of Rock-Water Interaction. Brantley, S.L.; Kubicki, J.D.; White, A.F., Eds. Springer: New York, 2008, pp. 39-72. • Leach, A. An Introduction to Computational Quantum Mechanics. In: Molecular Modeling Principles and Applications. 2nd Ed. Prentice Hall: U.K. 2001b, pp 65-74. • Lee, C.; Yang, W.; Parr, R.G Phys. Rev. B, 1988, 37, 785. • Martínez, C.; Rivera, J.L.; Herrera, R.; Rico, J.L.; Flores, N.; Rutiaga, J.G.; and López, P. J. Molec. Model. 2008, 14, 77-81. • Ralph, J.; Brunow, G.; Harris, P.J.; Dixon, R.A.; Schatz, P.F.; and Boerjan, W. Lignification: are Lignins Biosynthesized via simple Combinatorial Chemistry or via Proteinaceous Control and Template Replication? In Recent Advances in Polyphenol Research. Daayf, F. and Lattanzio, V., Eds. Blackwell: U.K., 2008a; Vol. 1; pp 36-66. • Sarotti, A.M; Pellegrinet, S.C. Journal of Organic Chemistry. 2009, 74, 7254-7260. • Szabo, A.; Ostlund, N.S. Model calculations on H2 and HeH+. In Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory. Dover Publications: Mineola, NY, 1996, pp 158. • Tossell, J.D. Calculating the NMR Properties of Minerals, Glasses, and Aqueous Species. In Molecular Modeling Theory: Applications in the Geosciences. Cygan, R.T. and Kubicki, J.D., Eds. Reviews in Mineralogy and Geochemistry: Washington, D.C., 2001; Volume 42; pp 437-458. • Watts, H.D.; Mohamed, M.N.A.; Kubicki, J.D. Journal of Physical Chemistry B. 2011, 115(9), 1958-1970.