Download

1 / 31
350 likes | 609 Views
Computational Techniques in Support of Drug Discovery. Jeffrey Wolbach, Ph. D. October 2, 2002. Who Is Tripos?. Discovery Software & Methods Research. Core Science & Technology. Software Consulting Services. Chemistry Products & Services. Discovery Research & Process Implementation.

E N D
Computational Techniques in Support of Drug Discovery Jeffrey Wolbach, Ph. D. October 2, 2002
Who Is Tripos? Discovery Software & Methods Research Core Science & Technology Software Consulting Services Chemistry Products & Services Discovery Research & Process Implementation
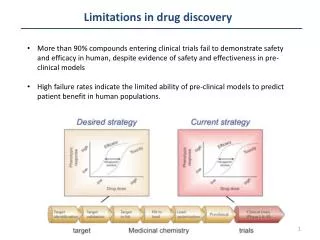
Choose Disease Target Identification Target Validation Lead Identification Lead Validation Lead Optimization Candidate to Clinic ADME Sequential Drug Discovery • Many cycles of synthesis/testing to identify and optimize lead • Role of molecular modeling • unrealistic to jump from validated target to optimized lead • useful to reduce the number of synthesis/testing cycles • enables “first to file” • enlarge number of targets
Choose a Disease Target Identification Target Validation Lead Identification Lead Validation Lead Optimization ADME Candidate to Clinic Drug Discovery in Parallel • Knowledge-sharing environment: genomics, HTS, chemistry, ADME, toxicology • Collect more data, on more compounds, more quickly • Apply predictive models of “developability” early • Enhanced understanding & predictive model building • Increase share of patented time on market
Ligand-Based Design Ligand Structures w/Activities No Target Structure Discern Similarities and Differences in Active Structures Pharmacophore Analysis QSAR Database Searching New Candidate Structures for Synthesis/Testing
Pharmacophore Analysis • Assume active molecules share a binding mode • Search for common chemical features of active molecules • Don’t know binding mode, so active molecules are considered flexible • Search set of pre-determined conformers • Allow molecules to flex during search • Typical features: • H-Bond Donors • H-bond acceptors • Hydrophobic groups
Pharmacophore Models • Chemical features in 3-D space • Distance constraints between chemical features
QSAR • Relates bioactivity differences to molecular structure differences • Structure represented by numerical descriptors • Traditional (2D) QSAR • 3D QSAR - CoMFA • Statistical techniques relate descriptors to activity + + + + + D Activity + + + + + + + Activity = D0 + 0.5 D1 + 0.17 D2 + ... D Descriptor
QSAR - Traditional (2D) • Descriptors are molecular properties • logP, dipole moment, connectivity indices ... Structures + Activity Descriptors Predictive Model (QSAR Equation) pKi=5.3 logP = 1.9 m = 2.8 Estate = 7.2 pKi=A + B(logP) + C(m) + D(Estate) + ... pKi=3.7 logP = 1.7 m = 2.3 Estate = 6.7 logP = 2.1 m = 3.5 Estate = 5.5 pKi=2.9 PLS MLR . .
QSAR - 3D QSAR - CoMFA • Comparative Molecular Field Analysis • Descriptors are field strengths around molecules - electrostatic, steric, H-bond .. • Fields can have easy physical interpretation pKi=A + B(D1) + C(D2) + ...
QSAR/CoMFA - Interpretation • High Coefficient (important) lattice points can be plotted around molecular structures
2D Database Searching 010110010010101 • Searches often performed on bit-strings • “Fingerprints” (many types) • Fingerprints display neighborhood behavior • Also includes substructure searching • Can search for similarity or dissimilarity
3D Database Searching • Query is a collection of features in 3-D space • Pharmacophore • Lead compound / specific atomic groups • Search a database of flexible, 3-D molecules • Molecules can’t be stored in every possible conformation • Allow molecules to flex to fit the query
Example of Structure-Based Design
3D Database Searching • Not restricted to ligand-based design • Information about target can be included in the query • Can define steric hindrances • Additional interaction sites • Serves to filter hits from the search
Identification of Novel Matrix Metalloproteinase (MMP) Inhibitors • MMPs • Zinc-dependent proteases • Involved in the degradation and remodeling of the extracellular matrix • They are important therapeutic targets with indications in: • Cancer • Arthritis • Autoimmunity • Cardiovascular disease A fibroblast collagenase-1 complexed with a diphenyl-ether sulphone-based hydroxamicacid
Objectives • Design high affinity MMP inhibitors based on the diketopiperazine scaffold by: • Creating a virtual combinatorial library of candidate inhibitors • Using virtual screening tools to identify candidates with the highest predicted affinity • Perform R-group and binding mode analysis to guide library design
Synthesis ofDKP-MMP inhibitors DKP-I DKP-II 1.) Esterification of the solid support (HO-) with an amino acid 2.) Reductive alkylation of the amino acid and acylation of the resulting secondary amine 3.) Deprotection of the N-alkylated dimer followed by cyclic cleavage from the resin yielding diketopiperazine (DKP)
Finding & Filtering Reagents UNITY 2D structure search of the ACD • Filtered out: • Metals • MW > 250 • RB > 8 • Filtered out: • Metals • MW > 400 • RB > 15 1154 aldehydes 73 Boc protected amino acids
Selecting Reagents & Building the Virtual Library • Selector™ • Diverse selection of • amino acids (R1) and • aldehydes (R2) using: • 2D Finger Prints • Atom Pairs • Hierarchical Clustering Legion™ Model the reaction and create virtual combinatorial library 55 amino acids (R1) x 95 aldehydes (R2) x 14 amino acids (R3) = 73,150 compounds (~75k, 8.5 MB) Randomly selected 14 amino acids for R3
The CombiFlexX Protocol • Select a diverse subset of compounds using OptiSim • Dock and score the compounds in the diverse subset using FlexX • Select unique core placements using OptiSim • Hold each core placement fixed in the binding site as each R-group is independently attached, docked, and scored. • Sum the scores of the "R-cores" and subtract the score of the common core • Computation times scale as the sum of the number of R-groups rather than as the product of the number of R-groups
Virtual Screeningof DKP-MMP Inhibitors • ~75k compound library • MMP target structure collagenase-1 (966c.pdb) • 150 diverse compounds selected and docked • 39 non-redundant core placements based on RMSD > 1.5 Å.
Virtual Screening Results • Docked 91% of the library • 36 compounds/minute • 331 compounds predicted to be more active than those published
Consensus Scoring Results • Extracted the top 1000 library compounds based on Flex-X score • Ranked the top library compounds and published “highly actives” using CScore • 10 compounds predicted by all scoring functions to be more active than “highly actives”
R-Group Analysis in HiVol Frequency of R-group use among 331 active virtual compounds R2 (95 reagents) R3 (14 reagents) R1 (55 reagents)
R-Group Analysis in HiVol, con’t. Frequency of R-group use among 331 active virtual compounds R1
Summary • Used CombiFlexX and HiVol to: • Identify highly promising candidates • Perform R-group analysis & Binding mode analysis to guide further computational design of libraries • Further Work • Diversity/similarity analysis of the published and virtual libraries • Use docking results for library design in Diverse Solutions • SAR development
Binding Mode Analysis Frequency of core placement use
R-Group Analysis in HiVol Frequency of R-group use among 331 active virtual compounds R2 (95 reagents) R3 (14 reagents) R1 (55 reagents)