Download

1 / 79
930 likes | 1.55k Views
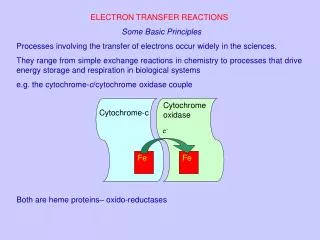
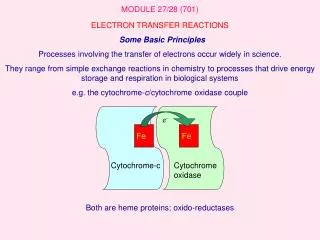
Electron Transfer Reactions. Electron transfer reactions occur by one of two fundamental mechanisms. In an inner sphere mechanism, there is a common bridging ligand, and the electron is transferred from the reductant to the oxidant through the bridging ligand.

E N D
Electron transfer reactions occur by one of two fundamental mechanisms In an inner sphere mechanism, there is a common bridging ligand, and the electron is transferred from the reductant to the oxidant through the bridging ligand
In an outer sphere mechanism, there is an encounter between the reductant and the oxidant. The electron is transferred from one to the other whilst there is no change in the coordination sphere of either.
Common bridging ligands Oxide and hydroxide. But water is a very poor bridging ligand
_ 2- Other examples Common bridging ligands Ligands which have more than one donor atom (called ambident nucleophiles)
Common bridging ligands Ligands which have more than one donor atom separated by a delocalised electron system
Henry Taube’s classic experiment [CoIII(NH3)5Cl]2+ Inert: d6 Co(III) [CrII(H2O)6]2+ Labile: d4 Cr(II) Observation: products (in acidic medium) are [CoII(H2O)6]2+ + [CrIII(H2O)5Cl]2+ How do we distinguish an inner sphere from an outer sphere mechanism? Cl- is a bridging ligand; neither H2O nor NH3 are and this allowed him to deduce the mechanism
electron transfer break apart hydrolysis, in acid [CoII(H2O)6]2+ + 5NH4+ The reaction could have occurred through an inner sphere pathway: [CoIII(NH3)5Cl]2+ + [CrII(H2O)6]2+ [CoIII(NH3)5ClCrII(H2O)5]4+ [CoII(NH3)5ClCrIII(H2O)5]4+ Co(II) is labile Cr(III) is inert [CoII(NH3)5(H2O)]2+ + [CrIII(H2O)5Cl]2+
electron transfer hydrolysis, in acid [CoII(H2O)6]2+ + 5NH4+ + Cl Or it could have gone through an outer sphere pathway: [CoIII(NH3)5Cl]2+ + [CrII(H2O)6]2+ Co(II) is labile Cr(III) is inert [CoII(NH3)5Cl]2+ + [CrIII(H2O)6]2+
Observation: products (in acidic medium) are [CoII(H2O)6]2+ + [CrIII(H2O)5Cl]2+ [CoII(H2O)6]2+ [CrIII(H2O)6]2+ So there would have had to be a subsequent anation of [CrIII(H2O)6]2+ by Cl Rate of [CrIII(H2O)6]2+ by Cl : k = 2.9 10-8 M-1 s-1 Rate of electron transfer:k = 6 10+5 M-1 s-1 Rate of electron transfer is 13 orders to magnitude faster than rate of anation
Rate of electron transfer is 13 orders to magnitude faster than rate of anation The reaction could not have proceeded through an outer sphere mechanism Taube’s postulate: A reaction will have proceeded through an inner sphere mechanism if one of the products is substitution inert and it retains the bridging ligand i.e., the rate of the electron transfer reaction is much faster than the rate of formation of the product by subsequent anation Corollary: If the rate of electron transfer is much faster than the rate of ligand substitution on either metal ion, the reaction must proceed through an outer sphere mechanism
Now [RuIII(NH3)5Br]2+ + [VII(H2O)6]2+ e transfer, k = 5.1 103 M-1 s-1 [RuII(NH3)5Br]+ + [VIII(H2O)6]3+ But [VII(H2O)6] + Br [VII(H2O)5Br]+ + H2O k = 5.0 101 s-1 Example V2+ is inert (d3 ion, high LFSE – like Cr3+) Ru3+ is inert (2nd transition series) Hence cannot form the inner sphere complex fast enough – the anation reaction is too slow. The reaction must have been outer sphere.
e- Donor Acceptor ΨDΨA Barriers to electron transfer • Distance Rate of an electronic transition HDA>2 where (Atkins, 8th ed., Chapter 9; 9th ed., Chapter 8) Hamiltonian operator that describes the coupling of the two wavefunctions
If the coupling is relatively weak, Electron coupling when A and D are in direct contact (r = 0) edge-to-edge distance between D and A Parameter that measures the sensitivity of the coupling to distance and it turns out that the rate constant for electron transfer between D and A is (Atkins, 8th ed., p. 897; not in 9th ed.)
A D ln kET r Will be a constant if D and A are the same
Nature often uses large, conjugated macrocycles to do electron transfer Examples: Chlorophylls Porphyrins
Effectively increases radius of D and A, cutting down separation, and hence increasing rate of e- transfer distance between metals effective distance
Q-cytochrome c oxidoreductase – Complex III or the bc1 complex
which is often written in simplified form as nuclear frequency factor electronic factor 0 ≤ κ ≤ 1 • For fast electron transfer, maximise κE
For fast electron transfer, maximise κE • minimise the reorganisation energy, λ, of inner and outer sphere
[Co(phen)3]3+ + [Co(bipy)3]2+ → [Co(phen)3]2+ + [Co(bipy)3]3+ low spin low spin low spin low spin eg t2g • use appropriate electronic configurations When an e- is transferred from the D to the A molecule, it cannot change its spin. In many cases this is not a problem:
[Co(NH3)4Cl2]3+ + [Co(OH2)6]2+ → [Co(NH3)4Cl2]2+ + [Co(OH2)6]3+ low spin high spin high spin low spin S = 0 S = 3/2 S = 3/2 S = 0 eg t2g This cannot be a single step But in some cases - especially if there is a change of spin state - this is a barrier to electron transfer
[Co(NH3)4Cl2]3+ + [Co(OH2)6]2+ → {[Co(NH3)4Cl2]3+}* + [Co(OH2)6]2+ low spin high spin excited state1 high spin eg t2g
{[Co(NH3)4Cl2]3+}* + [Co(OH2)6]2+→ {[Co(NH3)4Cl2]3+}* + [Co(OH2)6]2+ excited state1 high spin excited state2 high spin eg t2g
[Co(OH2)6]3+ and finally low spin {[Co(NH3)4Cl2]3+}* + [Co(OH2)6]2+→ [Co(NH3)4Cl2]2+ + [Co(OH2)6]3+ excited state2 high spin high spin excited state3 eg t2g
Faster electron transfer occurs if an electron is removed from and added to a non-bonding orbital (less reorganisational energy λ) Recall that in complexes with σ only ligands the t2g orbitals are non-bonding and eg orbitals are antibonding
Compare self-exchange rate constants: [Cr(OH2)6]3+/2+t2g3/t2g3eg1 1 × 10-5 M-1s-1 [Fe(OH2)6]3+/2+t2g3eg2/t2g4eg2 1.1 × 10-5 M-1s-1 [Ru(OH2)6]3+/2+ t2g5/t2g6 20 × 10-5 M-1s-1 electrons going in and out of the t2gorbitals makes for fast electron transfer
The Inner Sphere Mechanism • The rate-determining step could be • the formation of the bridged complex (i.e., the precursor complex) • the electron transfer step (most commonly rate determining) • the break-up of the successor complex
K [RuIII(NH3)5Cl]2+ + [CrII(H2O)6]2+ [RuIII(NH5)5ClCrII(H2O)5]4+ + H2O e transfer [RuII(NH5)5ClCrIII(H2O)5]4+ + H2O k1 [RuII(NH3)5(H2O)]2+ + [CrIII(H2O)5Cl]2+ We can often rationalise which step will be rate-determining Both Ru(II) and Cr(III) are inert – so we would expect the breakup of the successor complex to be rate-limiting
Changes in mechanism are often accompanied by substantial changes in rate The following reactions must be outer sphere reactions (why?) oxidant reductant k [CoIII(NH3)5(H2O)]3+ [RuII(NH3)6]2+ 3.0 M-1 s-1 [CoIII(NH3)5(OH)]2+ [RuII(NH3)6]2+ 0.04 M-1 s-1 and the hydroxo complex reacts some 100 times slower than the aqua complex oxidant reductant k [CoIII(NH3)5(H2O)]3+ [CrII(H2O)6]2+ 0.1 M-1 s-1 [CoIII(NH3)5(OH)]2+ [CrII(H2O)6]2+ 1.5 106 M-1 s-1 The hydoxo complex in this case must be reacting through an inner sphere mechanism
For the first row of the d block: • If electron transfer is rate-determining, then the rate depends markedly on 1. the identity of the metal ion These are the same factors that controlled rate in the outer sphere mechanism (see later) 2. the nature of the bridging ligands The ability of the ligand to act as an electron conductor
2. The ability of the ligand to act as an electron conductor para k = 100 M-1 s-1 k = 1.6 10-3 M-1 s-1 meta
[L5MoxX] + [L5MredY] rate limiting [L5Mox] + X + [L5MredY] [L5MoxYMredL5] [L5MredYMoxL5] etc If formation of the precursor complex is rate determining, then the rate is usually not very sensitive to the nature of the bridging ligand This is because the ligand substitution reactions of the first row d metals are usually dissociative hence does not depend strongly on the nature of the entering ligand Example: V2+(aq) is oxidised to V3+(aq) by a long series of Co3+ oxidants with different bridging ligands
Inner sphere mechanism always suspected if good bridging ligands are available: Cl- Br- I- N3- CN-
The Outer Sphere Mechanism Rudolph Marcus
Energy changes during electron transfer – the Frank-Condon Principle • Electron transfer is fast compared to nuclear motion • Hence the nuclei are essential frozen in space during the electron transfer step Now consider the following situation: [FeII(H2O)6]2+ + [*FeIII(H2O)6]3+ [FeIII(H2O)6]3+ + [*FeII(H2O)6]2+ Fe(II)-O = 2.02-2.07 Å Fe(III)-O = 2.00 Å
e Fe(III)OH2 + *Fe(II)OH2 bond too long for Fe(III) bond too short for Fe(II) getting energy from nothing – which would be a violation of the First Law spontaneous (exothermic) So suppose Fe(II)OH2 + *Fe(III)OH2 Fe(III)OH2 + *Fe(II)OH2
Fe(II)OH2 + *Fe(III)OH2 shrinks stretches ENDOTHERMIC Fe(II)OH2 + *Fe(III)OH2 bonds now about the same length G‡ e transfer rearrangement EXOTHERMIC Fe(III)OH2 + *Fe(II)OH2 What actually happens: Frank-Condon Energy Fe(III)OH2 + *Fe(II)OH2
A given system can (in principle) be represented by a wavefunction, Represents hypersurface of the reactants, reac represents changes to all structural parameters (bond lengths, angles, torsions, etc) during the reaction
Parabolic function because bond stretching and angle bending terms can be approximated by Hooke’s law behaviour: E0 x0 For example, [Fe3+(H2O)6] with a short Fe–O bond, xo
products For example, [Fe2+(H2O)6] with a long Fe–O bond
Electron transfer from the reactants to products can occur when the reactant deforms along the reaction coordinate until it structurally resembles the product (at ) For example, Fe3+––O must stretch
λis the reorganisation energy, the energy that would be expended to reorganise the reactant form to the product form if no electron transfer took place λ