Download

1 / 50
520 likes | 782 Views
B cell immunodeficiency Sara Mohammad Abuel Gassim November 2009.

E N D
B cell immunodeficiency Sara Mohammad Abuel Gassim November 2009
Introduction • B lymphocytes, named after their site of origin in the bursa of Fabricius in birds or in the bone marrow in humans, form the basis for humoral immunity by their production of immunoglobulins. • B-cell disorders are divided into defects of B-cell development/ immunoglobulin production (immunodeficiencies) and excessive/uncontrolled proliferation (lymphomas, leukemias).
Immuodeficiency • Immunodeficiency is the failure of the immune system to protect against disease or malignancy. • Primary Immunodeficiency is caused by genetic or developmental defects in the immune system. These defects are present at birth but may show up later on in life. • Secondary or acquired immunodeficiency is the loss of immune function as a result of exposure to disease agents, environmental factors, immunosuppression, or aging. • Bacterial, viral, protozoan, helminthic and fungal infections may lead to B cell, T cell, PMN and macrophage deficiencies. • Most prominent among these is acquired immunodeficiency syndrome (AIDS). • Secondary immunodeficiencies are also seen in malignancies.
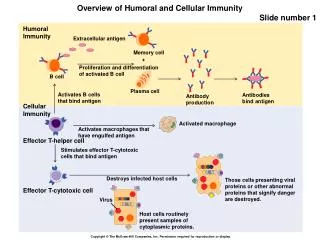
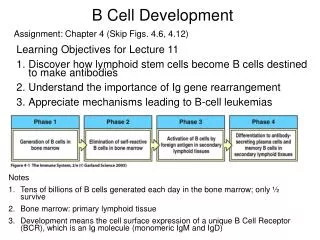
Pathophysiology • During fetal development, hematopoiesis including lymphopoiesis, is multicentric. • After birth, the bone marrow becomes the exclusive production site for lymphoid progenitors. • B and T cells, type 2 dendritic cells, and natural killer (NK) cells share a common ancestor, ie, common lymphoid progenitor (CLP). • CLP differentiates into 2 intermediate progenitors: early B cells and T/NK/dendritic trilineage cells. Both continue their development in the bone marrow through an antigen-independent process called primary lymphopoiesis (PL). • Recognized stages of PL are pro-B cell, pre-B cell, immature B cell, and mature B cell.
Secondary B lymphopoiesis is an antigen-dependent process and occurs in the germinal center of peripheral lymphoid organs with specific antibody production. • Secondary T lymphopoiesis is also an antigen-dependent process and occurs in the thymus. • Secondary lymphopoiesis (SL) begins when mature B cells enter the extrafollicular area of lymphoid tissue and differentiate into short-lived plasma cells and memory cells after first being stimulated by antigen-presenting cells.
Memory cells travel to the primary follicle, where, after exposure to dendritic cells, they differentiate into centroblasts (immunoglobulin class-switch). • Centroblasts progress to centrocytes with high-affinity antibody production, and then they differentiate further to long-term memory cells and plasmablasts. The latter migrate back to the bone marrow and start producing immunoglobulins.
The earlier the defect, the more devastating the effect on lymphopoiesis. • Defects occurring at the CLP stage or those affecting processes common to B- and T-cell development result in combined immunodeficiency involving B, T, and NK cells.
Primary B-cell disorders result in a complete or partial absence of one or more immunoglobulin isotypes. • Regardless of the primary cause, the symptoms depend on the type and severity of the immunoglobulin deficiency and the association of cell-mediated immunodeficiency. • In general, severe immunoglobulin deficiency results in recurrent infections with specific microorganisms in certain anatomical sites.
IgG deficiency • IgG deficiencies may be divided into 2 categories. The first is selective IgG deficiency, which consists of an isolated deficiency of IgG with normal levels of IgA, IgM, IgD, and IgE. The second is a deficiency of IgG accompanied by inadequate levels of other immunoglobulin isotypes. • This may occur in various conditions, including X-linked agammaglobulinemia (X-LA), common variable immunodeficiency (CVID), and hyper-IgM syndromes.
X-linked agammaglobulinemia(XLA) • Also known as Bruton agammaglobulinemia, is the result of a mutation of the BTK gene. Pro-B cells are present in normal number, but they are unable to mature to pre-B cells. • The BTK gene is present on Xq21.3-q22, and its defect results in deficiency of Bruton tyrosine kinase. • Non-XLA is the result of mu heavy-chain gene deficiency that leads to abortive production of IgM and failure of B-cell development.
Activated tyrosine kinases generate a second wave of messengers by activating serine/threonine kinases or phosphatases pathways. • Three major pathways have been identified: the inositol phospholipid hydrolysis pathway, the phosphatidyl inositol-3-kinase pathway, and the Ras pathway. • These pathways converge toward the activation of transcription factors, resulting in B-cell activation and proliferation.
Bruton’s Tyrosine Kinase • Signalling molecule B-cell development into anti-body producing plasma cells. • 5 domains all of which can be affected by disease causing alterations.
XLA – summary • Btk gene mutation. • Loss of functional tyrosine kinase. • Blocked B cell maturation.
Common Variable Immunodeficiency(CVID) • Mature B cells are normal in number and morphology but fail to differentiate to plasma cells because of defective interaction between T and B cells, mostly caused by a T-cell defect. This defect is thought to be related to a decreased number and/or function of CD4+ T lymphocytes or, occasionally, to an increased number of CD8+ T lymphocytes. • Abnormal responses of B cells to many usual stimuli have also been identified in vitro.
The underlying abnormality in selective IgM deficiency is a defect of helper T-cell and excessive suppressor T-cell activity. • The disorder is characterized by a low level of IgM. IgG levels are normal, but the IgG response is usually decreased. • Helper T-lymphocyte deficiency has been incriminated in the pathogenesis of transient hypogammaglobulinemia of infancy (THI) and immunodeficiency with thymoma. • The primary defect in selective IgA deficiency is related to a failure of B cells to differentiate to mature isotype-switched surface IgA-positive B cells and IgA-secreting plasma cells with appropriate stimuli. The basis for the defect is not known.
B cells in IgA deficiency activated via CD40 and interleukin-10 are capable of synthesizing and secreting IgA. Defective helper T-cell and excessive suppressor T-cell activities are occasionally present. • Cytokine abnormalities have also been described. • IgG2 is the most common of IgG subclass deficiencies. It occurs either alone or with IgG4 or IgA deficiency. Its hallmark is an inability to generate antibodies to polysaccharides.
Hyper-IgM immunodeficiency • Individuals with this type of immunodeficiency have low IgA and IgG concentrations with abnormally high levels of IgM. • These patients cannot make a switch from IgM to other classes which is attributed to a defect in CD40L on their CD4 cells. They are very susceptible to pyogenic infection and should be treated with intravenous gamma-globulins.
Thymoma • Some patients with thymomas have an associated IgG deficiency. In most cases, the thymoma is composed of a benign type of spindle cell, and both B- and T-cell deficiency have been reported. • Panhypogammaglobulinemia and deficient cell-mediated immunity usually occur. • This condition is usually referred to as Good syndrome. • Removal of the thymoma does not reverse the immunoglobulin deficiency, and • patients may benefit from IgG replacement therapy to prevent recurrent infection.
Transient hypogammaglobulinemia • Children, at birth, have IgG levels comparable to that of the mother. • Because the half life of IgG is about 30 days, its level gradually declines, but by three months of age normal infants begin to synthesize their own IgG. • In some infants, IgG synthesis may not begin until they are 2 to 3 years old. This delay has been attributed to poor T cell help. • This results in a transient deficiency of IgG which can be treated with gamma-globulin.
IgA deficieny • This is the most common antibody deficiency and results in chronic infections at mucosal surfaces since IgA is the secretory antibody which protects these surfaces. • Since IgG and IgM can compensate, to a significant degree, for the lack of IgA, this deficiency often goes unnoticed since it usually does not cause serious problems. • A few patients with IgA deficiency may also develop additional antibody deficiencies (for example deficiencies in IgG) and/or autoimmune disease with time.
X-linked immunodeficiency with hyper-IgM(XHM) • Is related to a deficiency in gp39 (CD40 ligand). • B cells can initiate the immune response by producing IgM, but they are not capable of operating the class-switching, hence the overproduction of IgM and the decrease or absence of the other immunoglobulin isotypes. • Liver disease, sclerosing cholangitis, and liver/GI malignancies are common in these patients.
The expression of CD40L on the surface of biliary epithelial cells has suggested a role for CD40-CD40L interaction in the pathogenesis of these complications through defective control of intracellular pathogens such as Cryptosporidium parvum, which has been recognized as an important pathogen in these patients.
Also known as acquired immunodeficiency. • one is born with normal immune responses but some secondary factor or occurrence causes a decrease in immune responses. • Secondary immunodeficiency is induced by factors such as:
Malnutrition • Most common cause of secondary immune deficiency • Protein-Calorie malnutrition can lead to abnormalities of T cells, B cells and phagocytes. • Atrophic and fibrotic thymus. • Reduced lymphocyte proliferation in response to antigens.
Malignancy • Immunodeficiency caused by lymphoid tumours e.g. leukaemia, lymphoma, plasma cell dyscrasias • Hodgkin’s lymphoma: causes classic example of T-cell deficiency. • Chronic lymphocytic leukaemia: causes classic example of B-cell deficiency. Lymphocyte Depletion Hodgkin's Lymphoma
Human Immunodeficiency Virus (HIV) • Human retrovirus • RNA genome. • Targets host cell immune system itself… “abnormalities in every arm of the immune system”
Other causes for secondary immunodeficiency: • Irradiation - exposure to X-rays and gamma rays. Causes a decreased production of lymphocyte precursors in the bone marrow. • Cytotoxic drugs such as many used in cancer chemotherapy. Causes a decreased production of lymphocyte precursors in the bone marrow. • Corticosteroids – anti-inflammatory steroids. Damages lymphocytes. • Aging. Adaptive immunity, especially cell-mediated immunity, tends to deminish with aging. • Removal of the spleen. Decreased ability to remove microbes that enter the blood.
Other clinical conditions • In studies performed to improve the accuracy of early diagnosis of retinoblastomas, markedly reduced blood and lacrimal fluid IgG levels have been found in persons with stage III-IV retinoblastoma. • A relationship between febrile convulsion and IgG subclass deficiencies has also been demonstrated. One study found that IgG subclass deficiencies may be responsible for an increased incidence of febrile infections, which result in a higher incidence of seizures in children who are predisposed to febrile seizures. • IgG2, IgG4, and IgA subclass deficiencies may play a role in the pathogenesis of allergic colitis in children.
Infants with a definitive diagnosis of noninfective colitis displayed a high prevalence of IgA, IgG2, and IgG4 deficiencies. • Although some reports have suggested a strong relationship between IgG subclass patterns and atopic disorders, other studies have not found this association.
T cell deficiencies with variable degrees of B cell deficiency
Ataxia-telangiectasia • Ataxia-telangiectasia is a deficiency of T cells associated with a lack of coordination of movement (ataxis) and dilation of small blood vessels of the facial area (telangiectasis). • T-cells and their functions are reduced to various degrees. • B cell numbers and IgM concentrations are normal to low. IgG is often reduced and IgA is considerably reduced (in 70% of the cases). • There is a high incidence of malignancy, particularly leukemias. • The defects arise from a breakage in chromosome 14 at the site of TCR and Ig heavy chain genes.
Wiskott-Aldrich syndrome (WAS) • This syndrome is associated with normal T cell numbers with reduced functions, which get progressively worse. IgM concentrations are reduced but IgG levels are normal. • Both IgA and IgE levels are elevated. Boys with this syndrome develop severe eczema, petechia (due to platelet defect and thrombocytopenia). • They respond poorly to polysaccharide antigens and are prone to pyogenic infection. • Wiskott-Aldrich syndrome is an X-linked disorder due to defect in a cytoskeletal glycoprotein, CD43.
Deficiencies in Cell Mediated Immunity • SCID • SCID refers to severe combined immune deficiency which results in no, or low numbers of, functional B and T cells. By far the most profound effect on the immune response is the lack of T cells. • Children with SCID are usually diagnosed in their first six months of life because of severe bacterial, viral and fungal infections. They must be kept isolated from any potential for infection. • This is the classic “baby in the bubble” syndrome.
The prognosis is very poor unless their immune system can be reconstituted by providing normal bone marrow stem cells through transplantation from an HLA-matched donor. • Since the development of B cells and T cells depends on a number of critical steps, all going according to plan, a defect in any of these critical steps will result in SCID.
A defect in the RAG genes which are required for gene rearrangement of both the T cell receptor and antibody will result in an inability of either T cells or B cells to reach maturity. • A common genetic defect leading to SCID is a problem in the gene coding for the gamma chain of the IL-2 receptor. This chain is also part of the receptors for IL-4, IL-7, IL-9 and IL-15. A defect in the gamma chain knocks out the activity of all of these important cytokines and renders the immune response completely ineffective. • A defect in the JAK kinase signal transduction pathway activated by these cytokines has a similar effect.