Download

1 / 54
540 likes | 781 Views
Alternative Splicing from ESTs. Eduardo Eyras Bioinformatics UPF – February 2004. Intro ESTs Prediction of Alternative Splicing from ESTs. Transcription. exons. introns. pre-mRNA. Splicing. Mature mRNA. Translation. Peptide. 5’. 3’. 3’. 5’. 5’ CAP. AAAAAAA. Different Splicing.

E N D
Alternative Splicing from ESTs Eduardo Eyras Bioinformatics UPF – February 2004
Intro • ESTs • Prediction of • Alternative Splicing from ESTs
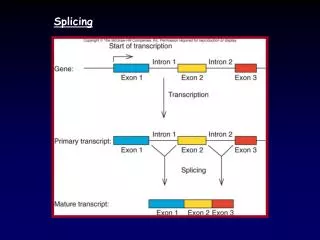
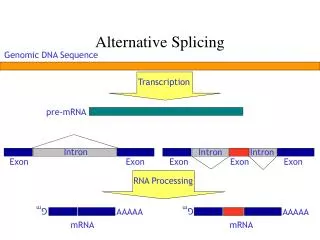
Transcription exons introns pre-mRNA Splicing Mature mRNA Translation Peptide 5’ 3’ 3’ 5’ 5’ CAP AAAAAAA
Different Splicing Mature mRNA Translation Different Peptide 5’ 3’ 3’ 5’ Transcription exons introns pre-mRNA 5’ CAP AAAAAAA
Alt splicing as a mechanism of gene regulation Functional domains can be added/subtracted protein diversity Can introduce early stop codons, resulting in truncated proteins or unstable mRNAs It can modify the activity of the transcription factors, affecting the expression of genes It is observed nearly in all metazoans Estimated to occur in 30%-60% of human
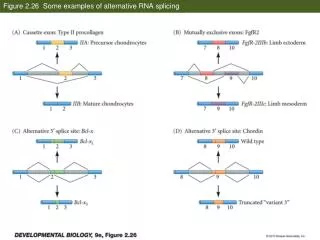
Forms of alternative splicing Exon skipping / inclusion Alternative 3’ splice site Alternative 5’ splice site Mutually exclusive exons Intron retention Constitutive exon Alternatively spliced exons
ESTs (Expressed Sequence Tags) Single-pass sequencing of a small (end) piece of cDNA Typically 200-500 nucleotides long It may contain coding and/or non-coding region
5’ 5’ 5’ 3’ 3’ 3’ AAAAAA AAAAAA AAAAAA ESTs Cells from a specific organ, tissue or developmental stage mRNA extraction Add oligo-dT primer TTTTTT 3’ 5’ Reverse transcriptase RNA TTTTTT DNA 3’ 5’ Ribonuclease H TTTTTT 3’ 5’ DNA polimerase Ribonuclease H 5’ 3’ AAAAAA Double stranded cDNA TTTTTT 3’ 5’
ESTs 5’ 3’ AAAAAA Clone cDNA into a vector TTTTTT 3’ 5’ 5’ EST Single-pass sequence reads Multiple cDNA clones 3’ EST
Sampling the Transcriptome with ESTs Genomic Primary transcript Splicing Splice variants oligo-dT primer Reverse transcriptase cDNA clones (double stranded) EST sequences (Single-pass sequence reads) 5’ 3’ 5’ 3’
EST sequencing • Is fast and cheap • Gives direct information about the gene sequence • Partial information Resulting ESTs Known gene (DB searches) Similar to known gene Contaminant Novel gene
dbEST release 20 February 2004 • Number of public entries: 20,039,613 • Summary by organism • Homo sapiens (human) 5,472,005 • Mus musculus + domesticus (mouse) 4,056,481 • Rattus sp. (rat) 583,841 • Triticum aestivum (wheat) 549,926 • Ciona intestinalis 492,511 • Gallus gallus (chicken) 460,385 • Danio rerio (zebrafish) 450,652 • Zea mays (maize) 391,417 • Xenopus laevis (African clawed frog) 359,901 • …
EST lengths ~ 450 bp Human EST length distribution (dbEST Sep. 2003 )
Anatomical System The tissue, organ or anatomical system from which the sample was prepared. Examples are digestive, lung and retina. Cell Type The precise cell type from which a sample was prepared. Examples are: B-lymphocyte, fibroblast and oocyte. Pathology The pathological state of the sample from which the sample was prepared.Examples are: normal, lymphoma, and congenital. Developmental Stage The stage during the organism's development at which the sample was prepared. Examples are: embryo, fetus, and adult. Pooling Indicates whether the tissue used to prepare the library was derived from single or multiple samples. Examples are pooled, pooled donor and pooled tissue. ESTs provide expression data eVOC Ontologieshttp://www.sanbi.ac.za/evoc/ J Kelso et al. Genome Research 2002
ESTs provide expression data eVOC Ontologieshttp://www.sanbi.ac.za/evoc/ Developmental Stage Anatomical System Pathology Cell Type Pooling … nervous brain cerebellum … Library 1 Library 2 … ESTs ESTs
Linking the expression vocabulary to gene annotations ESTs Genes V Curwen et al. Genome Research (2004)
The down side of the ESTs • Cannot detect lowly/rarely expressed genes or non-expressed sequences (regulatory) Random sampling: the more ESTs we sequence the less new useful sequences we will get
It defines the location of exons and introns We can verify the splice sites of introns check the correct strand of spliced ESTs It helps preventing chimeras It can avoid putting together ESTs from paralogous genes We can prevent including pseudogenes in our analysis ESTs aligned to the genome EST Stop * AG GT PolyA Processed pseudogene True match best in genome Paralog Must Clip poly A tails before aligning
Alternative Exons/ 3´ PolyA sites from ESTs ESTs can also provide information about potential alternative splicing when aligned to the genome (and when aligned to mRNA data)
Aligning ESTs to the Genome • Many ESTs Fast programs, Fast computers • Nearly exact matches Coverage >= 97% • Percent_id >= 97% • Splice sites: GT—AG, AT—AC, GC—AG
Genomics as a Technology Development of special software: fast versus accurate alignment Development of special technology: efficient use of computer farms (~2000 CPUs)
The Problem ESTs Genome What are the transcripts represented in this set of mapped ESTs?
Predict Transcripts from ESTs ESTs Transcript predictions Merge ESTs according to splicing structure compatibility
Redundant ESTs Consider 2 ESTs in a Genomic Cluster with more ESTS x z x + z z gives redundant splicing information, we could keep only x x z w x + z z + w However, the relation with other ESTs in the cluster is important: a third EST, w, is compatible with z but not with x. --> keep all relations
Extension of the exon structure Consider 2 ESTs in a Genomic Cluster with more ESTS x y x + y y extends x, we can assume that they are from the same mRNA x z w Our success will depend on the coverage of the exons. However, ESTs are 3’and 5’ biased (ESTs like z not so frequent), hence we will have fragmentation.
Representation For every 2 ESTs in a Genomic Cluster, we decide if they represent equivalent splicing structures The compatibility relation is a graph: x x Extension y y x Inclusion x z z E Eyras et al. Genome Research (2004)
Criteria of “merging” Allow edge-exon mismatches mismatches Allow internal mismatches Allow intron mismatches Is this intron real?
Transitivity x x y y Extension z w x Inclusion z x z w w This reduces the number of comparisons needed
ClusterMerge graph Each node defines an inclusion sub-tree y z y x z x Extensions form acyclic graphs x x y y z z w w E Eyras et al. Genome Research (2004)
Mergeable sets Example 1 2 3 4 5 6 7
Mergeable sets Example 1 3 1 2 3 2 5 7 4 5 6 4 6 7
Mergeable sets Example Root 1 3 1 2 3 2 5 7 4 5 6 4 6 7 Leaves
Mergeable sets Example Root 1 3 1 2 3 2 5 7 4 5 6 4 6 7 Leaves Lists produced: (1,2,3,5,6,7) ( 1,2,3,4,5,7)
Deriving the transcripts from the lists Internal Splice Sites: external coordinates of the 5’ and 3’ exons are not allowed to contribute
Deriving the transcripts from the lists Splice Sites: are set to the most common coordinate 5’ and 3’ coordinates: are set to the exon coordinate that extends the potential UTR the most
Single exon transcripts Reject resulting single exon transcripts when using ESTs
Alternative splicing • and comparative genomics
Conservation of Alternative Splicing Degree of conservation: 30-60% Methods: 1.- compare single events 2.- Cross-alignment of full transcripts
Exon Skipping Events Introns flanking alternatively spliced (skipped) exons have high sequence conservation. Higher on average than constitutive inrons. R Sorek & G Ast. Genome Research 13:1631-1637, 2003
Conserved Alternative Exon • Sequences regulating the (Alternative) splicing Flanking Introns Overrepresented hexamer (downstream) Overrepresented sequences in conserved introns (between human and mouse) may be Involved in the regulation of alternative splicing. Overrepresented: found in these introns more often than expected at random AND not found in intronic sequences flanking constitutive exons (and upstream of skipped ones) R Sorek & G Ast. Genome Research (2003) 13:1631-1637
Sequences regulating the (Alternative) splicing Conserved Alternative Exon Flanking Introns Overrepresented hexamer Not all types of events are equally conserved. Introns flanking alternative 5´and 3´exons, and retained introns, have higher sequence conservation. Sugnet CW, Kent WJ, Ares M Jr, Haussler D. Pac Symp Biocomput. 2004;:66-77
Frame preservation A Resch et al. Nucleic Acids Research 2004, 32 (4) 1261-1269