Download

1 / 73
770 likes | 1.28k Views
DIVISION OF ANTIVIRAL DRUG PRODUCTS’ ADVISORY COMMITTEE OCTOBER 3, 2001. TENOFOVIR DF. Advisory Committee Issues. Treatment indication Nonclinical and clinical assessment of the effects of Tenofovir DF on bone Analysis of resistance data Design of trials for traditional approval.

E N D
DIVISION OF ANTIVIRAL DRUG PRODUCTS’ ADVISORY COMMITTEE OCTOBER 3, 2001 TENOFOVIR DF
Advisory Committee Issues • Treatment indication • Nonclinical and clinical assessment of the effects of Tenofovir DF on bone • Analysis of resistance data • Design oftrials for traditional approval
Advisory Committee Issues Proposed treatment indication: VIREADTM , in combination with other antiretroviral agents, is indicated for the treatment of HIV-infected adults. This indication is based on analyses of plasma HIV-1 RNA levels and CD4 counts in two controlled trials of VIREADTM of 24 and 48 weeks duration in treatment experienced adults with evidence of HIV-1 replication despite ongoing antiretroviral therapy. At present, there are no results from controlled trials evaluating the effect of tenofovir on clinical progression of HIV.
Advisory Committee Issues • Treatment indication • Pivotal studies 902 and 907 were conducted in a treatment experienced adult population • on stable ARV therapy for at least 8 weeks • median duration of therapy of ~ 4 - 5 years • mean baseline viral load = 3.4 log10 • mean baseline CD4 counts = 410 cells/mm3 • baseline resistance mutations - NRTI(94%), PI(58%), NNRTI(~40%) • Requesting AC input regarding labeled indication
Advisory Committee Issues • Bone Effects • BMD reductions/osteomalacia were observed in 3 species • Mechanism not fully defined • Clinical trial data limited for BMD • Seeking advice regarding: • implications of nonclinical and clinical bone data • recommendations for additional studies • monitoring plans
Advisory Committee Issues • Virology data • VIREADTM NDA contains more virology data than other NDA • Many analyses evaluating HIV RNA response by baseline phenotype and genotype • Seeking AC comments on: • clinical resistance analyses • inclusion in product labeling
VIREADTM NDA • Submitted in May 2001 • submitted under accelerated approval regulations • for serious and life-threatening conditions • provide meaningful therapeutic benefit over existing therapies • drug has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit or on a clinical endpoint other than survival or irreversible morbidity • DAVDP requires 2 adequate and well-controlled trials of 24 weeks duration
Advisory Committee Issues • Traditional approval plans • Continued marketing is subject to the need to confirm findings to establish clinical benefit • DAVDP requires 2 studies of 48 weeks duration to support traditional approval • Study 903 is being conducted in naïve subjects and is fully enrolled • Compares tenofovir DF to stavudine on a background of lamivudine and efavirenz • Seeking AC advice regarding design of second study in pediatric population
Advisory Committee Agenda • 9:00 a.m. Gilead Presentation • 9:45 a.m. FDA Presentation • 10:30 a.m. Break • 10:45 a.m. Discussion • 12:00 p.m. Lunch • 1:00 p.m. Open Public Hearing • 2:00 p.m. Continue Discussion and Questions to the Committee • 5:00 p.m. Adjourn
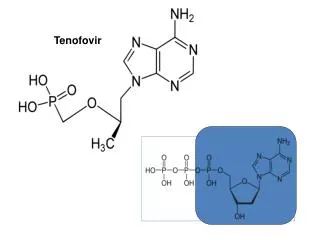
NDA 21-356Tenofovir Disoproxil Fumarate Kimberly Struble, PharmD Senior Regulatory Review Officer Division of Antiviral Drug Products
Presentation Outline • NDA Submission Overview • Efficacy Summary • Clinical Virology Results • Nonclinical Assessment of Bone Abnormalities - Jim Farrelly, Ph.D. • Clinical Assessment of Bone Abnormalities • Second Study for Traditional Approval • Summary of Regulatory Issues
NDA Overview • Submission Date: May 1, 2001 • Proposed Dosage: Tenofovir DF 300 mg once daily • Indication Sought: Treatment of HIV infection
NDA Submission:Four Clinical Studies • Supportive: • 901: Phase 2 dose finding (35 days) • 908: Compassionate Use Safety • Principal • 902 and 907: Randomized, Double-blind Placebo Controlled (24 weeks)
Principal Studies: 902 and 907 • Similar Study Designs • Safety and Efficacy of TNV vs PBO when added to stable ARV regimen in treatment experienced patients • Similar baseline characteristics • Differences in baseline HIV RNA • 902: Baseline HIV RNA 400-100,000 copies/mL • 907: Baseline HIV RNA 400-10,000 copies/mL
Primary Efficacy Endpoint • Primary endpoint = Time weighted change in log10 HIV RNA over 24 weeks (DAVG24) • DAVG is an acceptable endpoint for evaluating virologic responses in treatment experienced patients, such as those enrolled in 902 and 907 • Secondary endpoints = Proportion < 400 and 50 copies/mL
Mean Change From Baseline: HIV RNA DAVG24= - 0.61 DAVG24= - 0.58 0 2 4 8 12 16 20 24 WEEKS
Proportion < 400 and < 50 copies/mL Study 902 Study 907 Weeks Weeks
CD4 Response: Study 902 DAVG24= -3.6 DAVG24= -10.5 Weeks 4 8 12 24
CD4 Response: Study 907 DAVG24= +12.6 DAVG24= -10.6 Weeks 4 8 12 16 20 24
Efficacy Summary • Mean viral load reductions similar for 902 and 907 (Mean DAVG 0.5 - 0.6) • < 400 and < 50 copies/mL • numerical differences: 902 • statistically significant differences: 907 • Modest CD4 increases in study 907 • No differences for CD4 in study 902 over 24 weeks
Efficacy Summary (cont.) • Study population in 902 and 907 may not be optimal for observing large increases in CD4, given only one new drug added to stable regimen • Addition of one new agent did not produce substantial increases in CD4 over time • Further evaluations of CD4 in studies with different designs are needed
Clinical Virology • Applicant: HIV RNA response by prospectively defined baseline mutation subgroups • FDA: Exploratory analyses to further investigate HIV RNA response according to presence or absence of specific NRTI mutations • Determine if specific mutations or mutational patterns affected response to tenofovir
Clinical Virology: Limitations of FDA Analyses • Large number of potential comparisons limits ability to test for statistical significance • Limited # of patients for some primary NRTI and multi-drug resistant mutations to determine clinical significance • Given these limitations FDA is soliciting feedback on the types of exploratory analyses conducted and recommendations for labeling
Genotypic Results • HIV RNA response by presence or absence of thymidine analogue mutations (TAMs) • TAMs are defined as • M41L • D67N • K70R • L210W • T215Y/F • K219Q/E/N
HIV RNA Response by Baseline TAMs Mean DAVG24 (N) 67 + -0.53 (79) 67 - -0.62 (143) 70 + -0.71 (67) 70 - -0.54 (155) 219 + -0.60 (57) 219 - -0.58 (165)
HIV RNA Response by Baseline TAMs Mean DAVG24 (N) 215 - -0.80 (116) 215 + -0.35 (106) 210 - -0.70 (176) 210 + -0.17 (46) 41 - -0.78 (141) 41 + -0.26 (81)
Impact of 215 Mutation on HIV RNA Response Mean DAVG24 (N) 41 or 210 -0.25 (82) No 41 or 210 -0.70 (25) -0.80 (116) 215 -
Impact of 41 or 210 Mutation on HIV RNA Response Mean DAVG24 (N) No 41 or 210 -0.79 (139) -0.26 (93) 41 or 210
Other NRTI Mutations and HIV RNA Response • L74V/I affects tenofovir efficacy • DAVG24 = -0.17 (n=18) • Rates similar regardless if 41 or 210 mutation present with 74 (-0.12 to -0.19) • K65R mutation reduces susceptibility to tenofovir in vitro • DAVG24 = 0 (N=6) • More data needed to make any definitive conclusions
Phenotypic Analyses • To determine if tenofovir or other NRTI baseline susceptibility affected response • Baseline TNF susceptibility: • TNF < 4 fold • DAVG24 = -0.61 (n=91) • TNF > 4 fold • DAVG24 = -0.12 (N=9)
Resistance Summary • Genotypic data suggest potential for some cross resistance between tenofovir and specific NRTI mutations or patterns of mutations • However too few patients expressing some primary NRTI or multi-drug resistant NRTI mutations to determine clinical significance • No cross resistance between tenofovir and lamivudine
Resistance Summary • 41 or 210 mutation diminished responses, whereas 67, 70, 215 and 219 did not • Number and types of TAMs affect tenofovir efficacy • Efficacy reduced for > 3 TAMs which include M41L or L210W • K65R and L74V/I mutation may affect tenofovir efficacy • Reduced susceptibility to TNF (> 4 fold) at baseline diminishes tenofovir efficacy
Safety Summary • Treatment with TNF appears to be well tolerated • Most common AEs: asthenia (19%), headache (14%), diarrhea (22%), nausea (20%) and pharyngitis (18%) • GI events greater in TNF group vs PBO • diarrhea (22% vs 17%) • flatulence (6% vs 2%) • nausea (20% vs 15%) • vomiting (12% vs 6%)
Nonclinical Assessment of Bone Abnormalities James G. Farrelly, Ph.D. Pharmacology Supervisor
Toxicity in Rat and Dog Four Week Gavage Studies • Doses up to 500 mg/kg/day in rats • Little toxicity seen • Doses up to 30 mg/kg/day in dogs • Minor toxicity in kidney but no bone toxicity
Toxicity in Rat 42 Week Gavage Study • Doses @ 0, 30, 100, 300, 1000 mg/kg/d for 42 wks with 13 wk recovery and a 13 week interim evaluation • Bone Effects: • Bone mineral content and density • Cortical thickness of femur • Deoxypyridinoline at three highest doses • Osteocalcin at the two highest doses • Plasma phosphorus • Urinary calcium and phosphorus • PTH
Toxicity in Dog 42 Week Gavage Study • Doses @ 0, 3, 10, and 30 mg/kg/d for 42 wks with 13 wk recovery and a 13 week interim evaluation • Bone Effects • Bone mineral content and density • Urinary N-telopeptide • Urinary calcium and phosphorus • Bone specific ALP • 1,25-dihydroxy vitamin D3
Toxicity in Mouse 13 Week Gavage Study • Range-finding study to determine the maximum tolerated dose for a two year carcinogenicity study • Doses studied = 0, 100, 300, 600 mg/kg/d • Toxicity was seen in the kidney and duodenum • Carcinogenicity study is still ongoing
Intravenous Study in Cynomolgus Monkeys • Monkeys were dosed for 14 days by the intravenous route at doses up to 25 mg/kg/day with tenofovir • No bone toxicities were seen in this study • There were treatment findings in the kidneys
Rhesus Monkey Efficacy Studies • Studies designed to assess efficacy against SIV • Doses Studied = 10 and 30 mg/kg/day • Bone toxicities seen after 10 months dosing • Newbornmonkeys showed earlier bone toxicity when dosed at 30 mg/kg/day • At 10 mg/kg/day, no bone toxicity was seen in newborns dosed for two years
Rhesus Monkey Efficacy Studies, Cont. Bone Toxicity • Seen as abnormal growth plates and trabecula of femurs and ribs • Also seen were bone deformities and displacements, rib fractures and reduced bone density with bone loss • Reduction in serum phosphorus • Elevated ALP • Non-hyperglycemic glucosuria and proteinuria • Serum calcium unchanged
Rhesus Monkey Efficacy Studies, Cont. • It was concluded that treatment of rhesus monkeys at 30 mg/kg/day results in a mineralization defect in developing and growing cortical bone • The defect was considered to be osteomalacia • The defect was reversed by reducing the dose to 10 mg/kg/day or stopping treatment