Download
1 / 15
150 likes | 288 Views
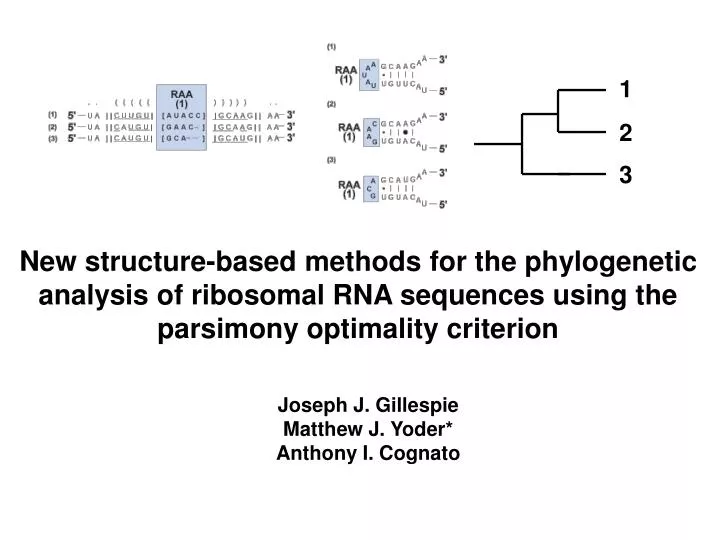
1. 2. 3. New structure-based methods for the phylogenetic analysis of ribosomal RNA sequences using the parsimony optimality criterion. Joseph J. Gillespie Matthew J. Yoder* Anthony I. Cognato. RNA molecules have characteristic higher order structure that is conserved across all life.

E N D
1 2 3 New structure-based methods for the phylogenetic analysis of ribosomal RNA sequences using the parsimony optimality criterion Joseph J. Gillespie Matthew J. Yoder* Anthony I. Cognato
RNA molecules have characteristic higher order structure that is conserved across all life Using structure to guide the assignment of positional nucleotide homology, we offer two new approaches to analyzing rRNA sequences: 1. RAA/RSC/REC coding 2. RNA basepair coding
RAA/RSC/REC coding* Methods for characterizing regions of RNA sequence alignments wherein positional nucleotide homology cannot be assigned with confidence Based on the premise of using information from secondary structure (i.e., compensatory base change evidence) to delimit unalignable positions *Gillespie (2004) Mol. Phylogenet. Evol. 33: 936-943
RAA/RSC/REC coding* RAA Region of ambiguous alignment Two or more adjacent, non-pairing positions within a sequence wherein positional homology cannot be confidently assigned due to the high occurrence of indels in other sequences *Gillespie (2004) Mol. Phylogenet. Evol. 33: 936-943
RAA/RSC/REC coding* RSC Region of slipped-strand compensation Region involved in base-pairing wherein positional homology cannot be defended across a multiple sequence alignment; inconsistency in pairing likely due to slipped-strand mispairing *Gillespie (2004) Mol. Phylogenet. Evol. 33: 936-943
RAA/RSC/REC coding* REC Region of expansion and contraction Variable helical region flanked by conserved basepairs at the 3’ and 5’ ends, and an unpaired terminal bulge of at least three nucleotides; characteristic of RNA hairpin-stem loops *Gillespie (2004) Mol. Phylogenet. Evol. 33: 936-943
RAA/RSC/REC coding* why? Subdividing large ambiguously aligned regions into smaller components provides: 1. a means for comparing structurally similar nucleotides in fragment level alignment methods (INAASE, POY) 2. fewer character state transformations between taxa, with less potential to exceed the number of allotted states in a given phylogenetic software 3. the ability to objectively assign different substitution weights to pairing (RSC, REC) and non-pairing (RAA) regions 4. improvements to existing global structural models for the various rRNA molecules on public databases 5. a more explicit set of homologies *Gillespie (2004) Mol. Phylogenet. Evol. 33: 936-943
RNA basepair coding code (20 states)* non-pairing pairing A = A AA = C CA = H GA = M UA = T C = R AC = Q CC = I GC = F UC = W G = N AG = E CG = L GG = P UG = Y U = D AU = G CU = K GU = S UU = V *adopted from Smith et al. (2004) Mol. Biol. Evol. 21: 419-427
RNA basepair coding substitution matrix non-pairing (-) canonical (-) non- canonical
RNA basepair coding weighting, i.e.
RNA basepair coding scripts available via the Jrna script package
http://hymenoptera.tamu.edu/rna This project was funded by NSF-PEET DEB grants 0328922 to Robert Wharton and 0358920 to Anthony Cognato.