Download

1 / 11
110 likes | 187 Views
Hierarchical Clustering in R. Quick R Tips. How to find out what packages are available library() How to find out what packages are actually installed locally (.packages()). Hierarchical Clustering. A type of cluster analysis

E N D
Quick R Tips • How to find out what packages are available • library() • How to find out what packages are actually installed locally • (.packages())
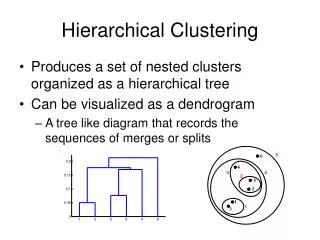
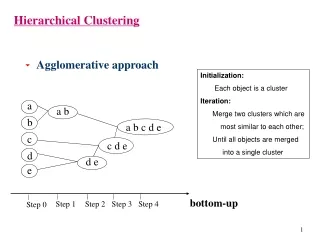
Hierarchical Clustering • A type of cluster analysis • There is both “divisive” and “agglomerative” HC…agglomerative is most commonly used • Group objects that are “close” to one another based on some distance/similarity metric • Clusters are created and linked based on a metric that evaluates the cluster-to-cluster distance • Results are displayed as a dendrogram
Step 1: Data matrix • First you need a numeric matrix • Typical array data set will have samples as columns and genes as rows • We want to be sure our data are in the form of an expression matrix • Use Biobase library/package • See http://www.bioconductor.org/packages/2.2/bioc/vignettes/Biobase/inst/doc/ExpressionSetIntroduction.pdf > exprs<-as.matrix(data, header=TRUE, sep="\t", row.names=1, as.is=TRUE)
Step 2: Calculate Distance Matrix • Default dist() method in R uses rows as the vectors..but we want the distance between samples….i.e., the columns of our matrix. • There is a handy package to help us at MD Anderson called oompaBase source("http://bioinformatics.mdanderson.org/OOMPA/oompaLite.R") oompaLite() oompainstall(groupName="all") • Once installed, be sure to locally activate the libraries library(oompaBase) library(ClassDiscovery) library(ClassComparison) • oompaBase also requires the mclust and cobs packages…download these from CRAN
Use the function distanceMatrix() to create a distance matrix of your samples…. • Uses the expression set created in Step 1 as input • Remember that there are many different types of distance metrics to choose from! • See help(distanceMatrix) x<- distanceMatrix(exprs,'pearson')
Step 3: Cluster • Use the hclust() function to create a hierarchical cluster based on your distance matrix, x, created in Step 2. > y<-hclust(x,method="complete") > plot(y)
Get the multtest package from CRAN • Package contains data from the Golub leukemia microarray data set (ALL v AML) • 38 arrays • 27 from lymphoblastic • 11 from myeloid http://people.cryst.bbk.ac.uk/wernisch/macourse/
library(multtest) • data(golub) • golub.cl • Generate the T statistic • teststat <-mt.teststat(golub, golub.cl) • Convert into P-values • rawp0 <-2*pt(abs(teststat),lower.tail=F, df=38-2) • Correct for multiple testing and show the ten most significant genes • procs <-c(“Bonferroni”, “BH”) • res<-mt.rawp2adjp((rawp0), procs) • res$adjp[1:10,] http://people.cryst.bbk.ac.uk/wernisch/macourse/