Download

1 / 1
10 likes | 166 Views
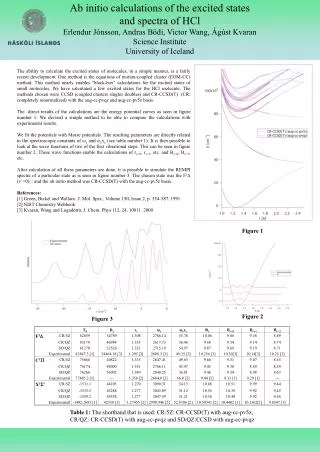
Ab Initio Study of the Hydrogenation of Mg with Incorporated Ti Atom on the Surface Aijun Du and Sean C. Smith Centre for Computational Molecular Science, Chemistry Building, The University of Queensland, Qld 4072. Introduction. Results & Discussions (continued).

E N D
Ab Initio Study of the Hydrogenation of Mg with Incorporated Ti Atom on the Surface Aijun Du and Sean C. Smith Centre for Computational Molecular Science, Chemistry Building, The University of Queensland, Qld 4072 Introduction Results & Discussions (continued) • Recent studies show that a single Ti atom coated on a single-walled nanotube (SWNT) and C60 fullerene could bind up to four hydrogen molecules [1-2]. • One Ti atom substituted one Mg atom on Mg(0001) surface could decrease the dissociation barrier of H2 to 0.10 eV [3]. • How about the interaction of many H2 molecules with Ti incorporated Mg(0001) surface? • The improved hydrogenation kinetics has experimentally been observed by mixing Ti, Carbon Nanotube and Mg materials during ball milling [4]. Figure 1a Figure 1b Computational Details • Ab initio PAW Method: All the calculations were performed using VASP code [5] implementing GGA of PBE exchange correlation functional. Projector Augmented Wave (PAW) method [6] is used to describe the electronic-ion-core interaction. Ti-incorporated Mg(0001) surface was modeled as a (3×3) surface unit cell with 5 layers of Mg atoms. Only gamma point calculation is performed. The vacuum space is at least 15 Å, which is enough to guarantee a sufficient separation between periodic images. • Nudged Elastic Band Method: The NEB method is used [7] to get Minimum Energy Path (MEP). A damped molecular dynamics was used to relax ions until the force in each image are less than 0.02 eV/Å. • Ab initio Molecular Dynamics Simulation: NVT ensemble (T=300K) with Nose thermostat. Deuterium is used instead of hydrogen during AIMD. MD time step is 0.5 fs. Figure 3 Energy profile for the dissociation of the second H2 molecule on Mg44TiH2 surface. The green, grey and pink balls represent Mg, Ti and H atoms, respectively. The activation barrier for the dissociation of the second H2 molecule on Ti-Mg(0001) surface (Mg44TiH2) is also small (0.145 eV) and we observe the spontaneous dissociation of second H2 on surface by using AIMD at 300K.. Results & Discussions The formation of Ti-incorporated Mg(0001) surface involves the creation of a Mg vacancy on Mg(0001) surface in the first step and the vacancy is then occupied by a Ti atom. Taking them together gives the adsorption energy (=1/9) at the surface substitutional site. The formation energy of Ti@Mg(0001) surface is calculated by Where ETi/Mg(0001)-subs, EMg, ETi-atom and EMg(0001) represent the total energy of the relaxed Ti-incorporated Mg(0001) surface, the bulk Mg atom, the isolated spin-polarized Ti atom and the clean Mg(0001) slab, respectively. The adsorption energy is calculated to be -4.09 eV, which indicated that Ti-incorporated Mg(0001) surface is thermodynamically stable. (a) (b) Figure 4 Final configuration for the third H2 molecule on Mg44TiH4 surface. Molecular adsorption state of H2 is first observed. The adsorption energy of hydrogen molecule is around 0.29 eV. • Polarization mechanism where the charge on the Ti cation polarizes the H2. • Further studies on the activation barrier for the diffusion of atomic H from surface to bulk show the macroscopically improvements in hydrogenation kinetics is still very limited. Other catalyst such as carbon is expected to be play an important role for in the atomic H diffusion process [8]. Conclusions Magnesium hydride (MgH2, 7.6 wt%) is a very promising approach for H2 energy carrier in mobile vehicles. Unfortunately, the application is primarily limited by the high hydrogenation reaction temperature and slow kinetics. In this work, we report new results from DFT studies of the dissociative chemisorption of molecular H2 on a Ti-incorporated Mg(0001) surface. It was found that two hydrogen molecules can dissociate on top of Ti atom with very small activation barrier (around 0.10 eV). Additionally, molecular adsorption of H2 on Ti atom on Mg(0001) surface is also observed. These results parallel recent findings for H2 adsorption on a Ti-decorated carbon nanotube [1] and clustered Ti on C60 fullerenes [2]. References [1 T.Yildirim and S. Ciraci, Phys.Rev.Letts., 94 (2005) 175501. [2] Qiang Sun, Qian Wang, Puru Jena and Yoshiyuki Kawazoe, J.Am.Chem.Soc, 127 (2005) 14582. [3] Aijun Du, S.C.Smith, X.D.Yao and G.Q.Lu, J.Phys.Chem.B, 109 (2005) 18037. [4] X. Yao, C. Z. Wu, Aijun Du, G. Q. Lu and Sean C. Smith J.Phys.Chem.B, (2006) in press. [5] Kresse, G.; Furthmuller, J. Comput. Mater. Sci1996, 6, 15. Kresse, G.; Furthmuller, J. Phys.Rev.B1996, 54, 11169. [6] Blochl, P.E. Phys.Rev.B1994, 50, 17953. Kresse, G.; Joubert, D. Phys.Rev.B1999, 59, 1758. [7] J.Henkelman and H.Jónsson, J.Chem.Phys., 2000, 113, 9978; 2000, 113, 9901. [8] Aijun Du,S.C.Smith, X.D.Yao and G.Q.Lu , J. Phys. Conference Series, 29 (2006)167. Figure 2 Energy profile for the dissociation of first H2 molecule on Ti-incorporated Mg(0001) surface. The black, grey and small white balls represent Mg, Ti and H atoms, respectively. IS, LS1, TS1, LS2 and FS represent the initial state, first local minimum state, first transition state, second local minimum state and final state, respectively. • It can be seen clearly from Figure 2 that there is a only a very small barrier for the dissociation of first H2 molecule on Ti-incorporated Mg(0001) surface. • AIMD at room temperature did observe the spontaneous dissociation of H2 on Ti-incorporated Mg(0001) surface. • Compared to pure Mg(0001) surface, the activation barrier of H2 decreased from 1.15 eV to 0.10 eV. • Strong interaction between the molecular orbital of H2 and the metal d orbital of Ti. Acknowledgements We acknowledge generous grants of high-performance computer time from both the Computational Molecular Science cluster computing facility at The University of Queensland and the Australian Partnership for Advanced Computing (APAC) National Facility. The authors also greatly appreciate the financial support by Australian Research Council through the ARC Center for Functional Nanomaterials.