Download

1 / 37
370 likes | 564 Views
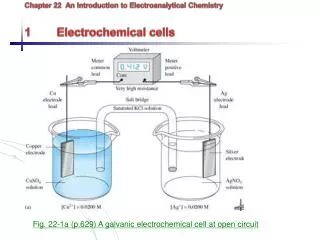
Electroanalytical Chemistry. Lecture #4 Why Electrons Transfer?. The Metal Electrode. E f = Fermi level; highest occupied electronic energy level in a metal. E F. E. E F. E redox. Why Electrons Transfer. Reduction. Oxidation. E redox. E. E. E F.

E N D
Electroanalytical Chemistry Lecture #4 Why Electrons Transfer?
The Metal Electrode • Ef = Fermi level; highest occupied electronic energy level in a metal EF E
EF Eredox Why Electrons Transfer Reduction Oxidation Eredox E E EF • Net flow of electrons from M to solute • Ef more negative than Eredox • more cathodic • more reducing • Net flow of electrons from solute to M • Ef more positive than Eredox • more anodic • more oxidizing
The Kinetics of Electron Transfer • Consider:O + ne- = R • Assume: • O and R are stable, soluble • Electrode of 3rd kind (i.e., inert) • no competing chemical reactions occur kR ko
Equilibrium for this Reaction is Characterised by... • The Nernst equation:Ecell = E0 - (RT/nF) ln (cR*/co*) • where: cR*= [R] in bulk solution co*= [O] in bulk solution • So, Ecell is related directly to [O] and [R]
Equilibrium (cont’d) • At equilibrium,no net current flows, i.e.,E = 0 i = 0 • However, there will be a dynamic equilibrium at electrode surface:O + ne- = RR - ne- = Oboth processes will occur at equal ratesso no net change in solution composition
Current Density, I • Since i is dependent on area of electrode, we “normalize currents and examineI = i/A we call this current density • So at equilibrium, I = 0 = iA + iC ia/A = -ic/A = IA = -Ic = Iowhich we call the exchange current density • Note: by convention iA produces positive current
Exchange Current Density • Significance? • Quantitative measure of amount of electron transfer activity at equilibrium • Io large much simultaneous ox/red electron transfer (ET) inherently fast ET (kinetics) • Io small little simultaneous ox/red electron transfer (ET) sluggish ET reaction (kinetics)
Summary: Equilibrium • Position of equilibrium characterized electrochemically by 2 parameters: • Eeqbm - equilibrium potential, Eo • Io - exchange current density
How Does I vary with E? • Let’s consider: • case 1: at equilibrium • case 2: at E more negative than Eeqbm • case 3: at E more positive than Eeqbm
Case 1: At Equilibrium E = Eo - (RT/nF)ln(CR*/CO*)E - E0 = - (RT/nF)ln(CR*/CO*) E = Eo so, CR* = Co*I = IA + IC = 0 no net current flows IA O G R IC Reaction Coordinate
Case 2: At E < Eeqbm • E - Eeqbm = negative number = - (RT/nF)ln(CR*/CO*) ln(CR*/CO*) is positive CR* > CO* some O converted to R net reduction passage of net reduction current IA O G R IC I = IA + IC < 0 Reaction Coordinate
Case 2: At E > Eeqbm • E - Eeqbm = positive number = - (RT/nF)ln(CR*/CO*) ln(CR*/CO*) is negative CR* < CO* some R converted to O net oxidation passage of net oxidation current IA R G IC O I = IA + IC > 0 Reaction Coordinate
Overpotential, fast slow • Fast ET = current rises almost vertically • Slow ET = need to go to very positive/negative potentials to produce significant current • Cost is measured in overpotential, = E - Eeqbm Cathodic Current, A Eeqbm Edecomp Cathodic Potential, V
Can We Eliminate ?What are the Sources of • = A + R + C • A, activationan inherently slow ET = rate determining step • R, resistancedue to finite conductivity in electrolyte solution or formation of insulating layer on electrode surface; use Luggin capillary • C, concentrationpolarization of electrode (short times, stirring)
Reference Working Electrode Luggin Capillary Luggin Capillary • Reference electrode placed in glass capillary containing test solution • Narrow end placed close to working electrode • Exact position determined experimentally
The Kinetics of ET • Let’s make 2 assumptions: • both ox/red reactions are first order • well-stirred solution (mass transport plays no role) • Then rate of reduction of O is: - kR co*where kR is electron transfer rate constant
The Kinetics of ET (cont’d) • Then the cathodic current density is: • IC = -nF (kRCO*) • Experimentally, kR is found to have an exponential (Arrhenius) potential dependence:kR = kOC exp (- CnF E/RT) • where C = cathodic transfer coefficient (symmetry) • kOC = rate constant for ET at E=0 (eqbm)
O G R Reaction Coordinate , Transfer Coefficient - measure of symmetry of activation energy barrier = 0.5 activated complex halfway between reagents/ products on reaction coordinate; typical case for ET at type III M electrode
The Kinetics of ET (cont/d) Substituting: • IC = - nF (kR co*) = = - nF c0*kOC exp(- CnF E/RT) Since oxidation also occurring simultaneously: • rate of oxidation = kA cR* • IA = (nF)kACR*
The Kinetics of ET (cont’d) • kA = kOA exp(+ AnF E/RT) • So, substitutingIA= nF CR*kOA exp(+AnF E/RT) • And, since I = IC + IA then: • I = -nF cO*kOC exp(- CnF E/RT) +nF cR*kOA exp(+ AnF E/RT)I = nF (-cO*kOC exp(- CnF E/RT) +cR*kOA exp(+ AnF E/RT))
The Kinetics of ET (cont’d) • At equilibrium (E=Eeqbm), recallIo = IA = - IC • So, the exchange current density is given by:nF cO*kOC exp(- CnF Eeqbm/RT) =nF cR*kOA exp(+ AnF Eeqbm/RT) = I0
The Kinetics of ET (cont’d) • We can further simplify this expression by introducing (= E + Eeqbm): • I = nF [-cO*kOC exp(- CnF ( + Eeqbm)/RT) + cR*kOA exp(+ AnF ( + Eeqbm)/ RT)] • Recall that ea+b = eaeb • So,I = nF [-cO*kOC exp(- CnF /RT) exp(- CnF Eeqbm/RT) + cR*kOA exp(+ AnF / RT) exp(+ AnF Eeqbm/ RT)]
The Kinetics of ET (cont’d) • So,I = nF [-cO*kOC exp(- CnF /RT) exp(- CnF Eeqbm/RT) + cR*kOA exp(+ AnF / RT) exp(+ AnF Eeqbm/ RT)] • And recall that IA = -IC = I0So,I = Io [-exp(- CnF /RT) + exp(+ AnF / RT)]This is the Butler-Volmer equation
The Butler-Volmer Equation • I = Io [- exp(- CnF /RT) + exp(+ AnF / RT)] • This equation says that I is a function of: • • I0 • C and A
The Butler-Volmer Equation (cont’d) • For simple ET, C + A = 1 ie., C =1 - A • Substituting:I = Io [-exp((A - 1)nF /RT) + exp(AnF / RT)]
Let’s Consider 2 Limiting Cases of B-V Equation • 1. low overpotentials, < 10 mV • 2. high overpotentials, > 52 mV
Case 1: Low Overpotential • Here we can use a Taylor expansion to represent ex:ex = 1 + x + ... • Ignoring higher order terms:I = Io [1+ (A nF /RT)- 1 - (A- 1)nF / RT)] = Io nF/RT • I = Io nF/RT so total current density varies linearly with near Eeqbm
Case 1: Low Overpotential (cont’d) • I = (Io nF/RT) intercept = 0slope = Io nF/RT • Note: F/RT = 38.92 V-1 at 25oC
Case 2: High Overpotential • Let’s look at what happens as becomes more negative then if IC >> IA • We can neglect IA term as rate of oxidation becomes negligible thenI = -IC = Io exp (-CnF /RT) • So, current density varies exponentially with
Case 2: High Overpotential (cont’d) • I = Io exp (-CnF /RT) • Taking ln of both sides:ln I = ln (-IC) = lnIo + (-CnF/RT) which has the form of equation of a line • We call this the cathodic Tafel equation • Note: same if more positive thenln I = ln Io + A nF/RT we call this the anodic Tafel equation
Tafel Equations • Taken together the equations form the basis for experimental determination of • Io • c • A • We call plots of ln i vs. are called Tafel plots • can calculate from slope and Io from y-intercept
Tafel Equations (cont’d) • Cathodic: ln I = lnIo + (-CnF/RT) y = b + m x • If C = A = 0.5 (normal), for n= 1 at RT slope = (120 mV)-1
Tafel Plots ln |i| High overpotential:ln I = lnIo + (AnF/RT) Anodic Cathodic Low overpotential:I = (Io nF/RT) ln Io Mass transport limited current _ Eeqbm + , V In real systems often see large negative deviations from linearity at high due to mass transfer limitations
EXAMPLE: • Can distinguish simultaneous vs. sequential ET using Tafel Plots • EX: Cu(II)/Cu in Na2SO4 • If Cu2+ + 2e- = Cu0 then slope = 1/60 mV • If Cu2+ + e- = Cu+ slow ?Cu+ + e- = Cu0 then slope = 1/120 mV • Reality: slope = 1/40 mVviewed as n = 1 + 0.5 = 1.5 • Interpreted as pre-equilibrium for 1st ETfollowed by 2nd ET
Effect of on Current Density • A = 0.75 oxidation is favored • C = 0.75 reduction is favored
Homework: • Consider what how a Tafel plot changes as the value of the transfer coefficient changes.