Download

1 / 17
170 likes | 312 Views
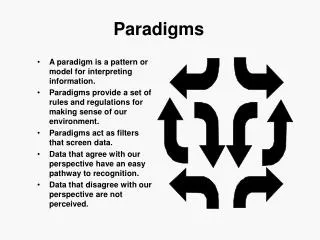
July 3, 2013 ONC HIT Health IT Policy Committee FDASIA WG. Event Reporting Paradigms. Slides for Discussion by FDASIA WG, Regulations Subcommittee Prepared by J. M. Goldman, MD, Co-Chair. V4. Contact: www.jgoldman.info. Data for Adverse Event Analysis.

E N D
July 3, 2013 ONC HIT Health IT Policy Committee FDASIA WG Event Reporting Paradigms Slides for Discussion by FDASIA WG, Regulations Subcommittee Prepared by J. M. Goldman, MD, Co-Chair V4 Contact: www.jgoldman.info
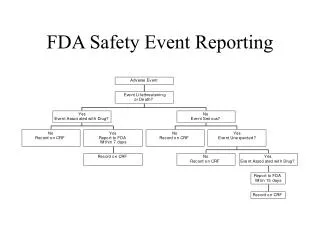
Data for Adverse Event Analysis • When medical device-HIT “system related” adverse events occur, it is often difficult or impossible to find the root cause of the failure. • The current HIT system does not lend itself to complete data logging for later analysis • Example: FDA ASTERD Pilot
FDA ASTERD • ASTERD - “ADE Spontaneous Triggered Event Reporting”* • The ASTER study was conceived as a proof of concept for a new model of gathering and reporting spontaneous Adverse Drug Events (ADEs). • Example: Ventilator – EHR can show when ventilator was exchanged (an unusual event) and prompt for more (manually entered) data. • But: Ventilator settings, error codes, patient physiological data, clinical impact, are not necessarily available, which limits the ability to analyze the events and mitigate future problems • Broader “system level” data logging is necessary (2) http://www.fda.gov/MedicalDevices/NewsEvents/WorkshopsConferences/ucm300722.htm http://www.fda.gov/downloads/MedicalDevices/NewsEvents/WorkshopsConferences/UCM329684.pdf
Wireless Infusion Pump Use Case 1/3 “An intravenous infusion pump is capable of securely being programmed via WiFi with a new drug library, or have the infusion rate adjusted for a closed-loop artificial pancreas function. Due to newly installed wireless equipment in proximity of the IV pump, WiFi commands to the pump are delayed (many minutes) or dropped, resulting in safety issues. The pump functions as ”specified". The manufacturer of the pump is contacted and states that the cause of the WiFi interference should be addressed.”
Simple Case: Pump produces smoke 2/3 • FDA MDR – Medical Device Reporting • “The Medical Device Reporting (MDR) regulation (21 CFR 803) contains mandatory requirements for manufacturers, importers and user facilities to report significant medical device adverse events to the Food and Drug Administration (FDA). FDA uses this information to identify and respond to problems associated with medical devices. • There is also a voluntaryMedWatch program for consumers or healthcare professionals to use to voluntarily report significant adverse events or product problems with medical products to FDA.” http://www.fda.gov/MedicalDevices/Safety/ReportaProblem/default.htm
Wireless Pump Use Case - continued 3/3 • Is this a single event? Endemic with this pump model? • What is the prevalence of this problem across all hospitals? • What is the clinical impact/severity? • Problem with this specific WiFi implementation? A signal of more general WiFi problems? • What if the data transmission to the EHR is lost? • What if data transmission is to another medical device? • Is newly installed technology (e.g. mBAN) causing interference? • How would this information be collected today (current state)? • Analyzed by whom? • Disseminated by whom? • Mitigations proposed and confirmed by whom? • Is that sufficient? • Future state?* *Supporting Medical Device Adverse Event Analysis in an Interoperable Clinical Environment: Design of a Data Logging and Playback System, International Conference on Biomedical Ontology: Workshop on Representing Adverse Events, 2011
FDA MedSun • The Medical Product Safety Network (MedSun) is an adverse event reporting program launched in 2002 by the U.S. Food and Drug Administration’s Center for Devices and Radiological Health (CDRH). • The primary goal for MedSun is to work collaboratively with the clinical community to identify, understand, and solve problems with the use of medical devices. • Participants use an Internet-based system that is designed to be an easy and secure way to report adverse medical device events. Each facility has online access to the reports they submit to MedSun so that they can be tracked and reviewed at any time. http://www.fda.gov/medicaldevices/safety/medsunmedicalproductsafetynetwork/default.htm
FCC § 15.5 General conditions of operation. (a) Persons operating intentional or unintentional radiators shall not be deemed to have any vested or recognizable right to continued use of any given frequency by virtue of prior registration or certification of equipment, or, for power line carrier systems, on the basis of prior notification of use pursuant to § 90.35(g) of this chapter. (b) Operation of an intentional, unintentional, or incidental radiator is subject to the conditions that no harmful interference is caused and that interference must be accepted that may be caused by the operation of an authorized radio station, by another intentional or unintentional radiator, by industrial, scientific and medical (ISM) equipment, or by an incidental radiator. (c) The operator of a radio frequency device shall be required to cease operating the device upon notification by a Commission representative that the device is causing harmful interference. Operation shall not resume until the condition causing the harmful interference has been corrected.(d) Intentional radiators that produce Class B emissions (damped wave) are prohibited. [54 FR 17714, Apr. 25, 1989, as amended at 75 FR 63031, Oct. 13, 2010]
FCC - continued · Unlicensed radio frequency devices such as digital electronic equipment (i.e., personal computers, e-readers, etc.) and low power radio transmitters such as Wi-Fi and Bluetooth are subject to the conditions that they do not cause harmful interference to authorized radio stations. The operator of the device must cease operation if notified by the FCC that the device is causing harmful interference. (See Section 15.5 of the FCC rules pasted below) · Licensed radio services (i.e., commercial cellular radio services, TV and radio broadcasting, etc.) generally operate on the principle that the most recently authorized services must avoid causing harmful interference to pre-existing services, and that having secondary frequency allocations may not cause harmful interference to services that have primary status. The FCC rules may be more specific about the responsibilities of that apply for any given service. In most cases, interference among licensed radio services is resolved directly by the licensees. · Most radio devices and transmitters must meet technical standards designed to minimize the risk of harmful interference are subject to equipment authorization requirements to ensure compliance. If an equipment manufacturer were to discover that its equipment is not in compliance with the standards or certification requirements it could be subject to FCC enforcement action. · In general there are no requirements to report harmful interference to the FCC, although here again there can be exceptions such as in instances where a device is authorized under a waiver with a condition that any harmful interference must be reported. However, the party receiving the interference could choose to file a complaint with the FCC which we would then investigate · FCC rules address interference to radio communications, not to devices such as electronic equipment that is not designed to transmit or receive on the airwaves.
EHR Configuration use case • “Assurance of Safe Configuration” • Safety of EHR depends on appropriate post-installation configuration. • No means to educate or require compliance with documented and evolving best practices
Support HIT Safety Framework: Is a system with feedback necessary? • Objective of system as whole – improve safety and efficiency of healthcare delivery* • Stakeholders need means (Data) to assess safety and performance of medical device – HIT system • Who are these stakeholders? • DeviceManufacturer • Health Delivery Organizations / Patients • Regulatory agencies • Payors? • Others? IOM 2011: Health IT and Patient Safety: Building Safer Systems for Better Care http://www.iom.edu/Reports/2011/Health-IT-and-Patient-Safety-Building-Safer-Systems-for-Better-Care.aspx
CPSC reporting • “If you are a manufacturer, importer, distributor, and/or retailer of consumer products, you have a legal obligation to immediately report the following types of information to the CPSC: a defective product that could create a substantial risk of injury to consumers or a product that is otherwise unreasonably hazardous or dangerous for consumers; a product that fails to comply with an applicable consumer product safety rule or with any other rule, regulation, standard, or ban under the CPSA or any other statute enforced by the CPSC; a product that a child (regardless of age) chokes on, such as a marble, small ball, balloon, or small part; and that, as a result of the incident, the child dies, suffers serious injury, ceases breathing for any length of time, or is treated by a medical professional; or certain types of lawsuits (applies to manufacturers and importers only, subject to the time periods detailed in Sec. 37 of the CPSA.) Failure to fully and immediately report this information may lead to substantial civil or criminal penalties.” http://www.cpsc.gov/Business--Manufacturing/Small-Business-Resources/Topics/Duty-to-Report-to-the-CPSC-Your-Rights-and-Responsibilities/
PSOs – Patient Safety Organizations • “The Patient Safety and Quality Improvement Act of 2005 (Patient Safety Act) authorized the creation of Patient Safety Organizations (PSOs) to improve the quality and safety of U.S. health care delivery. The Patient Safety Act encourages clinicians and health care organizations to voluntarily report and share quality and patient safety information without fear of legal discovery. • To implement the Patient Safety Act, the Department of Health and Human Services issued the Patient Safety and Quality Improvement Final Rule (Patient Safety Rule). The Agency for Healthcare Research and Quality (AHRQ) administers the provisions of the Patient Safety Act and the Patient Safety Rule dealing with PSO operations” http://www.pso.ahrq.gov
NHTSA “The mission of NHTSA is to “save lives, prevent injuries, reduce vehicle-related crashes”. To do so it must consider the effect on safety of factors as diverse as road surface composition, tire performance, signage, headlights, human factors, biomechanics, and, for when things don’t go well, crashworthiness, air bags, and child seats. Significant changes to the system must be evaluated in the context of all the other elements. NHTSA maintains data files and knowledge banks for research to improve vehicle safety, and has regulatory oversight.” Example: System perspective to safety http://www.nhtsa.gov/
ASRS • The ASRS is an important facet of the continuing effort by government, industry, and individuals to maintain and improve aviation safety. The ASRS collects voluntarily submitted aviation safety incident/situation reports from pilots, controllers, and others. • The ASRS acts on the information these reports contain. It identifies system deficiencies, and issues alerting messages to persons in a position to correct them. It educates through its newsletter CALLBACK, its journal ASRS Directline and through its research studies. Its database is a public repository which serves the FAA and NASA's needs and those of other organizations world-wide which are engaged in research and the promotion of safe flight. • [NTSB] http://asrs.arc.nasa.gov/
“HITSA” • Do we need a “Health IT Safety Administration” construct to address the gaps in reporting and analysis across regulated and non-regulated entities? • “Similarly, we could envision the creation of an “HIT Safety Administration” (HITSA) that could, in collaboration with vendors, hospitals, NIST, ONC and FDA provide a test bed for HIT systems prior to deployment and for solving problems when they develop. Proposed standards and technologies could be assessed and problems addressed prior to adoption (FCC, NSF, & NLM could provide expert input). Root cause analysis of adverse events and near misses could be performed in collaboration with vendors, as needed. Product defects or configuration problems could be addressed horizontally and solutions coordinated across industry, instead of by each hospital-vendor pair. Facilitation of adverse event analysis involving HIT systems may require the application of technologies that have been successful elsewhere, such as “black box” recorders for network medical device/HIT data.” *From briefing to Health Information Technology Innovation and Development Environment (HITIDE) group of the WH OSTP NITRD Health Information Technology Research and Development Senior Steering Group (HITR&D SSG) , Julian Goldman, MD, July 2011
Use Cases • Do we need to compile use cases that convey key ideas of • Scope of system • Stakeholders • Potential gaps in reporting, analyzing, mitigating hazards