Download

1 / 53
600 likes | 947 Views
Genome Assembly. Kelley Bullard, Henry Dewhurst, Kizee Etienne, Esha Jain, VivekSagar KR, Benjamin Metcalf, Raghav Sharma, Charles Wigington , Juliette Zerick. Outline. Stake Holders Biology NGS Review Introduction to Genome Assembly Challenges Analysis pipeline/ strategy

E N D
Genome Assembly Kelley Bullard, Henry Dewhurst, Kizee Etienne, Esha Jain, VivekSagar KR, Benjamin Metcalf, Raghav Sharma, Charles Wigington, Juliette Zerick
Outline • Stake Holders • Biology • NGS Review • Introduction to Genome Assembly • Challenges • Analysis pipeline/ strategy • Tool selection • Summary (final pipeline)
Stakeholders • CDC (Centers for Disease Control and Prevention) • GaTech • Immunocompromised individuals • Consumers of seafood • Prediction group (and subsequent groups) Stakeholders / Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Biology… Image of V. vulnificus Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Vibrio vulnificus • Gram-negative • Lipopolysaccharide membrane • Motile, facultative anaerobe • Halophilic (salt-loving) organism abundant in estuarine ecosystems • Major cause of seafood related deaths Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Vibrio vulnificus – genome architecture • Bacterial genomes are coding-dense • Introns rare • Contains plasmids (pYJ016) • V. vulnificus ~5.2mbp genome (similar to E. coli, ~50%) • GC content: 45-47% Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Vibrio navarrensis • Gram-negative • Lipopolysaccharide membrane • Motile, facultative anaerobe • Moderately halophilic organism • Some strains do not grow well in moderate to high salt concentrations Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Vibrio navarrensis- genomic architecture Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
NGS - Review Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Roche 454 sequencingworkflow overview Sample input: Genomic DNA, BACs, amplicons, cDNA Generation of small DNAfragments via shearing Ligation ofA/B-Adaptors flankingsingle- stranded DNAfragments Emulsification of beads and fragments in water-in-oil microreactors Clonal amplification of fragments bound to beads in microreactors Sequencing and base calling One Fragment One Bead One Read 400,000 reads per run Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
GSFLXData analysis – flowgramgeneration Flowgram Example of homopolymer errors from 454 sequencing data Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Example of 454 sff file (text format) Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Illuminasequencing overview 0.1 - 1.0μg cBot GAIIx User or corefacility Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Example of Illumina *.fastq file @C3PO_0001:2:1:17:1499#0/1 TGAATTCATTGACCATAACAATCATATGCATGATGCAAATTATAATATCATTTTTAGTGACGTCGTGAATCGTTT +C3PO_0001:2:1:17:1499#0/1 abaaaaaaaaaaa`a`aa_aaaaaaaaaaaaaaaa_aaaa`aaaaa^aaaaa`a]^`aYZYZ^`NJDJ\_Z @C3PO_0001:2:1:17:1291#0/1 TGTTTGAGCAAATGATTCATAATAATGTATTTCAATATTTTTAGGAATATCTCCCAATATTGCGCGTGCTGAATT +C3PO_0001:2:1:17:1291#0/1 a`_`_\a_aaaa_a^Z^^a[a^aa]a_^_a_``aa`aa`X^X^^`aa_\_]VR`\a_]W\_`_a]a]][\RZV @C3PO_0001:2:2:1452:1316#0/1 GTCCATCCGCAGCAGCGAATTTTTGACGTCCCCCCCCGAANGGANGNGANNNNGNNGNNNTNTNNAAANGNNNNN +C3PO_0001:2:2:1452:1316#0/1 _Ua\`]_`ZP\\_Z^[]aa^a_]XNBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBBB … Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Input reads Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
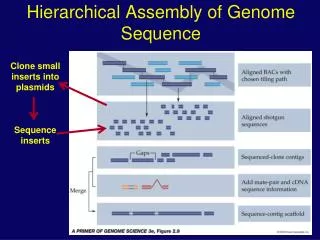
Introduction to genome assembly • An assembly is a hierarchical data structure that maps the sequence data to a putative reconstruction of the target. • In addition to contigs, a set of unassembled or partially assembled reads is also given as an output. Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
How do we check the quality of our assembly? METRICS! • N50 • minimum/maximum contig length • No. of contigs • No. of errors • FRC (feature response curve) Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Feature-by-Feature – evaluating de-novo assembly • BREAKPOINT: Points in the assembly where leftover reads partially align; • COMPRESSION: Area representing a possible repeat col- lapse; • STRETCH: Area representing a possible repeat expansion; • LOW_GOOD_CVG: Area composed of paired reads at the right distance and with the right orientation but at low coverage; • HIGH_NORMAL_CVG: Area composed of normal oriented reads but at high coverage; • HIGH_LINKING_CVG: Area composed of reads with associated mates in another scaffold; • HIGH_SPANNING_CVG: Area composed of reads with associated mates in another contig; • HIGH_OUTIE_CVG: Area composed of incorrectly oriented mates (--> -->, <-- -->); • HIGH_SINGLEMATE_CVG: Area composed of single reads (mate not present anywhere); • HIGH_READ_COVERAGE: Region in assembly with unexpectedly high local read coverage; • HIGH_SNP: SNP with high coverage; • KMER_COV: Problematic k-mer distribution. Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Feature-by-Feature – evaluating de-novo assembly • Most of the traditional metrics used to evaluate assemblies (N50, mean contig size, etc.) emphasize only size, while nothing (or almost nothing) is said about how correct the assemblies are. • A typical such metric (especially, in the NGS context) consists in aligning contigs back to an available reference. However, this naive technique simply counts the number of mis-assemblies without attempting to distinguish or categorize them any further. • After running amosvalidate, each contig is assigned the number of features that correspond to doubtful sequences in the assembly. • For a fixed feature threshold w, the contigs are sorted by size and, starting from the longest, only those contigs are tallied, if their sum of features is ƒw. For this set of contigs, the corresponding approximate genome coverage is computed, leading to a single point of the Feature-Response curve (FRC). Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Assembly Challenges Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Challenges • Intrinsic • Genome architecture • Repeats • Homopolymer runs • Sequence complexity • Chimeras? • Contaminants • Technical • Short reads • Poisson distribution of coverage • Sequencing errors • Variable quality • Sequence tags
454 • Illumina DeNovo • Allpaths LG • SOAP DeNovo • Velvet • Abyss • Taipan • Bambus2 • SUTTA • Hybrid DeNovo • Ray • MIRA Parameter optimization 454 raw reads Illumina Illumina raw reads hybrid • 454 DeNovo • Newbler • CABOG • SUTTA Process Illumina 454 • GAGE Statistical analysis Pre-processing Info. Evaluation Assemblers Illumina/ 454/ Hybrid DeNovo assembly • GAGE • Hawk-eye • Fastqc • Prinseq • NGS QC Assemblers Chosen Ref. Unmapped reads All possible combinations of the best 3 LEGEND 454 reads Illumina reads Read stats contigs * 3 • Mimimus • MAIA Finished genome Pre-processing Scaffolds Align illumina reads against 454 contigs Contig merging Unmapped reads • DNA Diff • PAGIT • Mauve • Mac vector • CLC wb Published Genomes from public databases V. vulnificus YJ016 V. vulnificus CMCP6 V. vulnificus MO6-24/O contigs Gap filling Nulceotide identity DeNovo assembly Genome finishing • bwa • GRASS • Built-in Unmapped reads Align Illumina against the reference • samstats contigs Compare mapping statistics Reference genome Illumina/(454?) reference based assembly Draft/ Finished genome • DNA Diff • DNA Diff • AMOScmp Reference evaluation Reference evaluation Reference selection Reference based assembly Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges / Analysis Pipeline-Strategy / Tool Selection / Summary
Tool Selection - Assembly Algorithm profile Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Greedy Seed-and-extension Graph based Branch-and-Bound • Basic operation: given any read or contig, add one more read or contig until no more reads or contigs are available • The contigs grow by “greedy extension” always incorporating a read that is found with the highest scoring overlap • Makes locally optimal choice with the hope of finding a globally optimal choice • No foresight -> misassembly Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Greedy Seed-and-extension Graph based Branch-and-Bound It was the best of age of wisdom, it was best of times, it was it was the age of it was the age of It was the best of it was the worst of was the best of times, the best of times, it of times, it was the best of times, it was of times, it was the of times, it was the of wisdom, it was the of times, it was the the age of wisdom, it times, it was the worst the best of times, it times, it was the age the worst of times, it times, it was the age times, it was the worst was the age of wisdom, • It was the best of times,it was the [worst/age] was the age of foolishness, was the best of times, was the worst of times, Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Greedy Seed-and-Extension Graph based Branch-and-Bound • Variation of the greedy assembler • Common in aligners, thus some assemblers/aligners may incorporate this approach • Particularly designed for short reads based on a contig heuristic scheme • Prefix-tree data structure • A contig is elongated at either end contingent upon the existence of reads with a prefix of minimal length perfectly matching the end of the contig Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Greedy Seed-and-extension Graph based Branch-and-Bound Overlap-layout-consensus (OLC): pairwise consensus Overlap: find potentially overlapping reads Layout: layout the reads based on matching alignment Consensus: derive the DNA sequence consensus by joining read sequences ..ACGATTACAATAGGTT.. Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Greedy Seed-and-extension Graph based Branch-and-Bound HamiltonianApproach Find an assembled sequence that explains the observed sequence = finding a path through a graph that visits every vertex once Repeat Repeat Repeat Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Greedy Seed-and-extension Graph based Branch-and-Bound de-Brujin Graph PotentialGenomesAAGACTCCGACTGGGACTTT AAGACTGGGACTCCGACTTT • Basic operation: k-mer approach • Eulerian approach de Bruijn Graph CCG TCC Reads AAGA ACTT ACTC ACTG AGAG CCGA CGAC CTCC CTGG CTTT … CGA CTC AAG AGA GAC ACT CTT TTT GGA CTG GGG TGG Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Greedy Seed-and-extension Graph based Branch-and-Bound • Basic operation: relies on “consistent layouts”; it generates all possible consistent layouts organizing them as paths in a “double tree” structure, rooted at a randomly selected seed read • Progressive evaluation of optimal criteria encoded by a set of score functions based on the set of overlaps along the layout Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Tid-bits of advice Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Tools of Choice Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
454 platform assembly Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Evaluation of 454 assemblers • Genomes Used For Comparison Comparative analysis of algorithms for whole-genome assembly of pyrosequencing data Brief Bioinform (2012) 13(3): 269-280 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of 454 assemblers using E. coli genome Comparative analysis of algorithms for whole-genome assembly of pyrosequencing data Brief Bioinform (2012) 13(3): 269-280 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of 454 assemblers using E. coli genome • The maximum value reached by the bars is the hypothetical reconstruction HR, defined as the ratio between the assembled bases and the reference length • The white section represents the real reconstruction RR, i.e. the portion of genome correctly reconstructed by assemblers. • The difference between hypothetical and RR, here called erroneous reconstruction ER, is shown in black Comparative analysis of algorithms for whole-genome assembly of pyrosequencing data Brief Bioinform (2012) 13(3): 269-280 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Illuminaplatform assembly Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Evaluation of illumina assemblers • Genomes Used For Comparison GAGE: A critical evaluation of genome assemblies and assembly algorithms. Steven L. Salzberg, Adam M. Phillippy, Aleksey Zimin, et al. Genome Res. 2012 22: 557-567 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of illumina assemblers • The best value for each column is shown in bold. For all assemblies • The Errors column contains the number of misjoins plus indel errors >5 bp for contigs, and the total number of misjoins for scaffolds. • Corrected N50 values were computed after correcting contigs and scaffolds by breaking them at each error. See the evaluation section in the text for details on how errors were identified. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Steven L. Salzberg, Adam M. Phillippy, Aleksey Zimin, et al. Genome Res. 2012 22: 557-567 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of illumina assemblers • A ‘‘chaff’’ contig is defined as a single contig <200 bp in length. In many cases, these contigs can be as small as the k-mer size used to build the de Bruijn graph (e.g., 36 bp) and are too short to support any further genomic analysis. • A duplicated repeat is one that appears in more copies than necessary in the assembly, and a compressed repeat is one that occurs in fewer copies. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Steven L. Salzberg, Adam M. Phillippy, Aleksey Zimin, et al. Genome Res. 2012 22: 557-567 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of illumina assemblers • ‘‘Misjoin’’ errors are perhaps the most harmful type, in that they represent a significant structural error. A misjoin occurs when an assembler incorrectly joins two distant loci of the genome, which most often occurs within a repeat sequence. • We have tallied three types of misjoins: (1) inversions, where part of a contig or scaffold is reversed with respect to the true genome; (2) relocations, or rearrangements that move a contig or scaffold within a chro- mosome; and (3) translocations, or rearrangements between chromosomes GAGE: A critical evaluation of genome assemblies and assembly algorithms. Steven L. Salzberg, Adam M. Phillippy, Aleksey Zimin, et al. Genome Res. 2012 22: 557-567 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of illumina assemblers • Average contig (A) and scaffold (B) sizes, measured by N50 values, versus error rates, averaged over all three genomes for which the true assembly is known: S. aureus, R. sphaeroides, and human chromosome 14. • Errors (vertical axis) are measured as the average distance between errors, in kilobases. • In both plots, the best assemblers appear in the upper right. GAGE: A critical evaluation of genome assemblies and assembly algorithms. Steven L. Salzberg, Adam M. Phillippy, Aleksey Zimin, et al. Genome Res. 2012 22: 557-567 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Applicability of assemblers • Genomes used for comparison A Practical Comparison of De novo Genome Assembly Software Tools for Next-Generation Sequencing Technologies.Wenyu Zhang, et al. Plos One. 2011 6: 1-12 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of illumina assemblers A Practical Comparison of De novo Genome Assembly Software Tools for Next-Generation Sequencing Technologies.Wenyu Zhang, et al. Plos One. 2011 6: 1-12 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Comparison of illumina assemblers A Practical Comparison of De novo Genome Assembly Software Tools for Next-Generation Sequencing Technologies.Wenyu Zhang, et al. Plos One. 2011 6: 1-12 Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Hybrid Platform Assembly Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Feature-by-Feature – evaluating de-novo assembly • COMPRESSION: Area representing a possible repeat col- lapse; • LOW_GOOD_CVG: Area composed of paired reads at the right distance and with the right orientation but at low coverage; • HIGH_OUTIE_CVG: Area composed of incorrectly oriented mates (--> -->, <-- -->); • HIGH_SINGLEMATE_CVG: Area composed of single reads (mate not present anywhere); • HIGH_READ_COVERAGE: Region in assembly with unexpectedly high local read coverage; • KMER_COV: Problematic k-mer distribution. Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Feature-by-Feature: evaluating de-novo assembly • Real Data - Long Reads Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary
Feature-by-Feature – evaluating de-novo assembly • Real Data - Short Reads Stakeholders/ Biology / NGS Review / Introduction to Genome Assembly / Challenges /Analysis Pipeline-Strategy / Tool Selection / Summary