Download

1 / 20
200 likes | 332 Views
Finding Regulatory Signals in Genomes. The Computational Problem Non-homologous/homologous sequences Known/unknown signal 1 common signal/complex signals/additional information Combinations. Regulatory signals know from molecular biology Different Kinds of Signals

E N D
Finding Regulatory Signals in Genomes The Computational Problem Non-homologous/homologous sequences Known/unknown signal 1 common signal/complex signals/additional information Combinations Regulatory signals know from molecular biology Different Kinds of Signals Promotors Enhancers Splicing Signals a-globins in humans
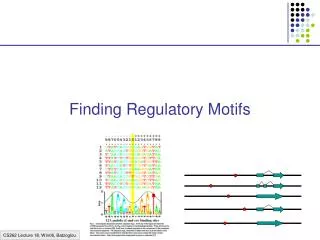
1 2 3 4 5 6 7 8 9 10 11 12 13 14 1 G A C C A A A T A A G G C A 2 G A C C A A A T A A G G C A 3 T G A C T A T A A A A G G A 4 T G A C T A T A A A A G G A 5 T G C C A A A A G T G G T C 6 C A A C T A T C T T G G G C 7 C A A C T A T C T T G G G C 8 C T C C T T A C A T G G G C Set of signal sequences: Consensus sequence: B R M C W A W H R W G G B M A 0 4 4 0 3 7 4 3 5 4 2 0 0 4 C 3 0 4 8 0 0 0 3 0 0 0 0 0 4 G 2 3 0 0 0 0 0 0 1 0 6 8 5 0 T 3 1 0 0 5 1 4 2 2 4 0 0 1 0 Position Frequency Matrix - PFM Position Weight Matrix - PWM A -1.93 .79 .79 -1.93 .45 1.50 .79 .45 1.07 .79 .0 -1.93 -1.93 .79 C .45 -1.93 .79 1.68 -1.93 -1.93 -1.93 .45 -1.93 -1.93 -1.93 -1.93 .0 .79 G .0 .45 -1.93 -1.93 -1.93 -1.93 -1.93 -1.93 .66 -1.93 1.3 1.68 1.07 -1.93 T .15 .66 -1.93 -1.93 1.07 .66 .79 .0 .79 -1.93 -1.93 -1.93 .66 -1.93 1 2 3 4 5 6 7 8 9 10 11 12 13 14 Score for New Sequence T T G C A T A A G T A G T C .45 -.66 .79 1.66 .45 -.66 .79 .45 -.66 .79 .0 1.68 -.66 .79 Sequence Logo & Information content Weight Matrices & Sequence Logos Wasserman and Sandelin (2004) ‘Applied Bioinformatics for the Identification of Regulatory Elements” Nature Review Genetics 5.4.276
1 (R,l) K 1.0 0.0 Motifs in Biological Sequences 1990 Lawrence & Reilly “An Expectation Maximisation (EM) Algorithm for the identification and Characterization of Common Sites in Unaligned Biopolymer Sequences Proteins 7.41-51. 1992 Cardon and Stormo Expectation Maximisation Algorithm for Identifying Protein-binding sites with variable lengths from Unaligned DNA Fragments L.Mol.Biol. 223.159-170 1993 Lawrence… Liu “Detecting subtle sequence signals: a Gibbs sampling strategy for multiple alignment” Science 262, 208-214. Q=(q1,A,…,qw,T) probability of different bases in the window A=(a1,..,aK) – positions of the windows q0=(qA,..,qT) – background frequencies of nucleotides. Priors A has uniform prior Qj has Dirichlet(N0a) prior – a base frequency in genome. N0 is pseudocounts
The Gibbs Sampler Target Distribution is Both random & systematic scan algorithms leaves the true distribution invariant. The conditional distributions are then: An example: x2 x1 The approximating distribution after t steps of a systematic GS will be: For i=1,..,d: Draw xi(t+1) from conditional distribution p(.|x[-i](t)) and leave remaining components unchanged, i.e. x[-i] (t+1) = x[-i](t)
Objective: Find conserved segment of length k in n unrelated sequences 1 2 n Remove one at random - sj 1 1 1 k k k (q1,..qk) Form profile of remaining n-1 Let pi be the probability with which sj[i..i+k-1] fits profile. Including pseudo-counts. Choose to start replacement at i with probability proportional to pi The Gibbs sampler Gibbs iteration: From : Lawrence, C. et al.(1993) Detecting Subtle Sequence Signals: A Gibbs Sampler approach to Multiple Alignment. Science 262.208-
Observed pattern in aligned sequences • Pattern Probability Model • Upper Points: original sequences • Middle: original date minus pattern • Lower: Shuffled sequences • 3 independent runs • a run on sequences without signal The Gibbs sampler: example From : Lawrence, C. et al.(1993) Detecting Subtle Sequence Signals: A Gibbs Sampler approach to Multiple Alignment. Science 262.208-
Multiple Pattern Occurances in the same sequences: Liu, J. `The collapsed Gibbs sampler with applications to a gene regulation problem," Journal of the American Statistical Association 89 958-966. Prior: any position i has a small probability p to start a binding site: width = w length nL ak Composite Patterns: BioOptimizer: the Bayesian Scoring Function Approach to Motif Discovery Bioinformatics Natural Extensions to Basic Model I Modified from Liu
Correlated in Nucleotide Occurrence in Motif: Modeling within-motif dependence for transcription factor binding site predictions. Bioinformatics, 6, 909-916. Insertion-Deletion BALSA: Bayesian algorithm for local sequence alignment Nucl. Acids Res.,30 1268-77. Start M2 w1 1 w2 w3 M3 K M1 w4 Regulatory Modules: De novo cis-regulatory module elicitation for eukaryotic genomes. Proc Nat’l Acad Sci USA, 102, 7079-84 Stop Gene A Gene B Natural Extensions to Basic Model II
Motifs Coding regions Expresssion and Motif Regression: Integrating Motif Discovery and Expression Analysis Proc.Natl.Acad.Sci. 100.3339-44 1.Rank genes by E=log2(expression fold change) 2.Find “many” (hundreds) candidate motifs 3.For each motif pattern m, compute the vector Sm of matching scores for genes with the pattern 4.Regress E on Sm ChIP-on-chip - 1-2 kb information on protein/DNA interaction: An Algorithm for Finding Protein-DNA Interaction Sites with Applications to Chromatin Immunoprecipitation Microarray ExperimentsNature Biotechnology, 20, 835-39 Protein binding in neighborhood Coding regions Combining Signals and other Data Modified from Liu
j Zi,j = 1 if a motif starts at j’th position in i’th sequence, otherwise 0. i 1 2 3 n 1 1 1 1 1 1 k k k k k k MEME- Multiple EM for Motif Elicitation Motif nucleotide distribution: M[p,q], where p - position, q-nucleotide. Background distribution B[q], l is probability that a Zi,j = 1 Find M,B, l,Z that maximize Pr (X, Z | M, B, l) Expectation Maximization to find a local maximum Iteration t: Expectation-step: Z(t) = E (Z | X, (M, B, l) (t)) Maximization-step: Find (M, B, l) (t+1) that maximizesPr (X, Z(t)| (M, B, l) (t+1)) Bailey, T. L. and C. Elkan (1994). "Fitting a mixture model by expectation maximization to discover motifs in biopolymers." Proc Int Conf Intell Syst Mol Biol 2: 28-36.
Phylogenetic Footprinting (homologous detection) Term originated in 1988 in Tagle et al. Blanchette et al.: For unaligned sequences related by phylogenetic tree, find all segments of length k with a history costing less than d. Motif loss an option. begin signal end Blanchette and Tompa (2003) “FootPrinter: a program designed for phylogenetic footprinting” NAR 31.13.3840-
The Basics of Footprinting I • Many aligned sequences related by a known phylogeny: positions HMM: 1 n 1 sequences k slow - rs HMM: fast - rf • Two un-aligned sequences: A C G T ATG A-C A
Statistical Alignment andFootprinting. • Many un-aligned sequences related by a known phylogeny: acgtttgaaccgag---- 1 • Conceptually simple, computationally hard • Dependent on a single alignment/no measure of uncertainty acgtttgaaccgag---- sequences sequences 1 k k acgtttgaaccgag---- 1 sequences Alignment HMM k Alignment HMM Signal HMM Solution: Cartesian Product of HMMs
Structure HMM S F F F S S 0.1 0.1 0.1 0.1 0.9 0.9 F S SF FS SS FF (A,S) F F S S F Alignment HMM ? Structure HMM “Structure” does not stem from an evolutionary model • The equilibrium annotation • does not follow a Markov Chain: • Each alignment in from theAlignment HMM • is annotated by the Structure HMM: • No ideal way of simulating: using the HMM at the alignment will give other distributions on the leaves using the HMM at the root will give other distributions on the leaves
Unrelated genes - similar expression Related genes - similar expression gene promotor Combine above approaches:Mixed genes - similar expression Combine “profiles” (Homologous + Non-homologous) detection Wang and Stormo (2003) “Combining phylogenetic data with co-regulated genes to identify regulatory motifs” Bioinformatics 19.18.2369-80
Regulatory Signals in Humans Transcription in Eukaryotes is done by RNA Polymerase II. 1850 DNA-binding proteins in the human genome. • Transcription Start Site - TSS • Core Promoter - within 100 bp of TSS • Proximal Promoter Elements - 1kb TSS • Locus Control Region - LCR • Insulator • Silencer • Enhancer Sourece: Transcriptional Regulatory Elements in the Human GenomeGlenn A. Maston, Sara K. Evans, Michael R. GreenAnnual Review of Genomics and Human Genetics. Volume 7, Sep 2006
Core Promoter Elements TATA - box Inhibitor element (Inr) Downstream Promoter Element (DPE) Downstream Core Element (DCE) TFIIB-recognition element (BRE) Motif Ten Element (MTE) Examples of Disease Mutations: Core promoter b-thallasemia TATA-box b-globin Enhancer X-linked deafness 900kb deletion POU3P4 Silencer Asthma 509bp mut TSS TFG-b Activator Prostate Cancer ATBF1 Coactivator Parkinson disease DJ-1 Chromatin Cancer BRG1/BRM Sourece: Transcriptional Regulatory Elements in the Human GenomeGlenn A. Maston, Sara K. Evans, Michael R. GreenAnnual Review of Genomics and Human Genetics. Volume 7, Sep 2006
a-globins Multispecies Conserved Sequences - MCSs Analyzed 238kb in 22 species Found 24 MCSs Programs use GUMBY - VISTA - MULTIPIPMAKER MULTILAGAN - CLUSTALW - DIALIGN TRANSFAC 6.0 - TRES - Experimental Knowledge of the region Hypersensitive sites (DHSs) DNA Methylation Region lies in CG rich, gene rich region close to the telomeres. It is not easy to align CG-islands.
Promoters in a-globins • 94.273-114.273 vista illus. • 5 MCSs • Divergence relative to human Promoters MCSs - 11 Regulatory MCSs - 4 Intronics MCSs - 2 Exonic MCSs - 4 Unknown - 3 Sourece: Hughes et al.(2005) Annotation of cis-regulatory elements by identification, subclassification, and functional assessment of multispecies conserved sequences PNAS 2005 102: 9830-9835;
Regulatory Protein-DNA Complexes Luscome et al.(2000) An overview of the structure of protein-DNA complexes Genome Biology 1.1.1-37 Moses et al.(2003) “Position specific variation in the rate fo evolution of transcription binding sites” BMC Evolutionary Biology 3.19- • Databases with the 3-D structure of combined DNA -Protein • Data bases with known promoters