Download
1 / 14
200 likes | 546 Views
Contact Order and Protein Folding Kinetics. K Plaxco et al (1998), J Mol Biol , 227:985-994. D Baker (2000), Nature, 405:39-42. Topic 16. Protein folding. Can we use structural bioinformatics to tell us anything about protein folding?. Two-state protein folding.

E N D
Contact Order and Protein Folding Kinetics K Plaxco et al (1998), J MolBiol, 227:985-994. D Baker (2000), Nature, 405:39-42. Topic 16
Protein folding Can we use structural bioinformatics to tell us anything about protein folding?
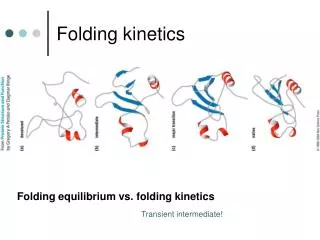
Two-state protein folding Cooperativity is a hallmark of protein structure and function. Ea U F N
Contact Order Relative CO is the average sequence distance between all pairs of contacting residues normalized by the total sequence length. N is the total number of contacts L is the total number of residues in the protein DSij is the sequence separation (in residues) between contacting residues i & j
Contact Order The basic idea is that it would take structural contacts that are separated far apart in sequence longer to form than structural contacts that are sequence neighbors. Low contact order (Faster folder) High contact order (Slower folder)
CO webserver http://depts.washington.edu/bakerpg/contact_order/
Such a simple idea… …has spawned myriad “Me too!” reports. 1, |i - j| > 12 0, otherwise Where nij = Meaning it gives the average number of structural contacts separated by 12 or more sequence positions.
Yet another CO variant… Istomin, Jacobs, and Livesay (2007). Protein Sci, 16:2564-2569.
Long-range order From the abstract: By analyzing correlation of other topological parameters with folding rates of two-state proteins, we find that only the long-range order exhibits correlation with folding rates that is uniform over all three classes. It is also the only descriptor to provide statistically significant correlations for each of the three structural classes. Istomin, Jacobs, and Livesay (2007). Protein Sci, 16:2564-2569.
Evolutionary Optimization of Protein Folding Our results show a clear overall increase of folding speed during evolution, with known ultra-fast downhill folders appearing rather late in the timeline. Debes et al. (2013). PLoS Computational biology 9(1):e1002861.
Evolutionary Optimization of Protein Folding Our results show a clear overall increase of folding speed during evolution, with known ultra-fast downhill folders appearing rather late in the timeline. Using phylogenomic and structural analyses, we observe an overall decrease in folding times between 3.8 and 1.5 billion years ago, which can be interpreted as an evolutionary optimization for rapid folding. Debes et al. (2013). PLoS Computational biology 9(1):e1002861.
Evolutionary Optimization of Protein Folding Our results show a clear overall increase of folding speed during evolution, with known ultra-fast downhill folders appearing rather late in the timeline. Using phylogenomic and structural analyses, we observe an overall decrease in folding times between 3.8 and 1.5 billion years ago, which can be interpreted as an evolutionary optimization for rapid folding. In contrast, we observed an increase in SMCO between 1.5 Gya and the present. Thus, the appearance of many new structures by domain rearrangement 1.5 Gya, also referred to as the “big bang” of the protein world, affected the evolutionary optimization of protein folding. Debes et al. (2013). PLoS Computational biology 9(1):e1002861.