Download

1 / 22
220 likes | 422 Views
Allineamenti e ricerca in Banche Dati: BLAST. Allineamenti. Per fare un allineamento ci vogliono:. >seq1 VATTDVADHFAC >seq2 GRAATQIRVLW. Le sequenze, e una matrice di sostituzione, che per ogni coppia di elementi (aa o basi) mi dà un punteggio, più

E N D
Allineamenti • Per fare un allineamento ci vogliono: >seq1 VATTDVADHFAC >seq2 GRAATQIRVLW Le sequenze, e una matrice di sostituzione, che per ogni coppia di elementi (aa o basi) mi dà un punteggio, più un punteggio da associare a inserzioni o cancellazioni (“trattini” o per meglio dire “gap” nell’allineamento)
Allineamenti • Una volta deciso se lo vogliamo locale o globale, il “programma” (o meglio, l’algoritmo) mi allinea le sequenze in modo da produrre il punteggio più alto possibile calcolato sommando colonna per colonna il punteggio associato a ciascuna coppia di elementi allineati
Allineamenti • Ma, sarebbe bello potere fare anche qualcosa di più • Finora abbiamo ipotizzato che le sequenze da confrontare fossero già note - le avessimo già scelte secondo qualche criterio: è possibile trovare “automaticamente” sequenze che si allineano “bene” con una sequenza data?
Allineamenti • Ma, sarebbe bello potere fare anche qualcosa di più • E’ possibile trovare un criterio “automatico” (al di là del colpo d’occhio) che ci possa dire con ragionevole affidabilità se un allineamento può essere considerato “buono”, ovvero sintomatico di un legame evolutivo (o funzionale) tra le sequenze confrontate? • …e l’allineamento multiplo? Come si fa a calcolarlo, e che informazioni fornisce?
Ricerche per Similarità • Possiamo utilizzare appositi programmi per chiedere: esiste (depositata in banca dati da qualcuno) una sequenza “sufficientemente” simile alla mia? • E: quando posso dire che due sequenze sono “sufficientemente” simili??
Ricerche per Similarità • Abbiamo un sistema per misurare la similarità: l’allineamento - che a ogni coppia di sequenze associa un punteggio • Quindi, data una sequenza di partenza, possiamo allinearla “contro” tutte le sequenze della banca dati, e fare una “classifica” sulla base del punteggio di similarità • Normalmente, l’evoluzione procede a blocchi: quindi, viene utilizzato normalmente l’allineamento locale
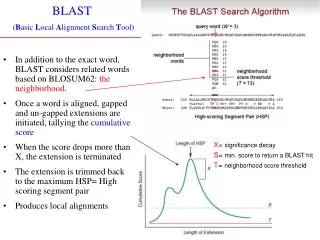
BLAST • BLAST (che sta per Basic Local Alignment Search Tool) è lo standard “de facto” per ricerche di questo tipo • Normalmente, ogni banca dati ha “incorporata” una ricerca per similarità tramite BLAST • … e, in effetti BLAT (Blast-Like Alignment Tool) è una versione semplificata e specializzata per allineare una sequenza ad un genoma • Noi utilizzeremo il BLAST associato alla banca dati “principale” (l’NCBI)
Blocchi conservati • Così • ... e non così
B LAS T a o l e o s c i a o i a g r l c l m c e h n t
Riassunto • E’ possibile eseguire una “ricerca per similarità” in una banca dati di sequenze • Ovvero, data una sequenza di partenza (“query”) si misura con l’allineamento locale la similarità della sequenza con tutte quelle della banca dati • Lo strumento più usato si chiama BLAST, e oltre a fare la classifica delle sequenze del database sulla base della similarità con la query, dà anche un’idea della “significatività” della similarità riscontrata
BLAST L’utilizzo è semplice: si inserisce la sequenza, e si sceglie “contro” quale insieme di sequenze allinearla. Normalmente, gli altri parametri sono impostati automaticamente
BLAST • ..e quindi, data la sequenza di input (detta “query”), il programma: • La allinea con ciascuna delle sequenze del database del tipo che avete selezionato • Riporta la “classifica” dei migliori allineamenti (o per lo meno i primi classificati) sulla base del punteggio… ma non solo:
Blast Output (finale) Punteggio allineamento difficile dire se “433” è buono oppure no… MA: Quanti allineamenti così “buoni” mi aspetto per caso?
10 alla meno... • 2 E -20 si legge come: 2 x 10 (-20)
Blast Output (in fondo...) Allineamenti “probabilmente” frutto del caso
Un po’ di Statistica • E-value (valore atteso) • Numero di sequenze del database che possono produrre per caso un allineamento con il punteggio trovato: più è piccolo, più è improbabile che si tratti di un “match” casuale • Se l’E-value è “quasi uno”, allora vuol dire che un allineamento con quel punteggio può essere trovato semplicemente perché in banca dati ci sono milioni di sequenze
Blast Output (per ogni sequenza “pescata” dal database) Locale! Non “copre” tutta la sequenza!
BLAST E-Value • E-value (o valore atteso) è il numero (atteso) di sequenze che mi aspetto di trovare per caso in banca dati tali da produrre un allineamento con punteggio maggiore o uguale a quello dato • Maggiore è il punteggio, minore è l’E-value • Minore è l’E-value, più è probabile che l’allineamento non sia casuale
Vari BLAST • L’algoritmo è sempre quello: ci sono però “adattamenti” a seconda del tipo di sequenza query e del database in cui si fa la ricerca: • BLASTn : query nucleotidi, database nucleotidi • BLASTp : query proteina, database proteine • BLASTx: query nucleotidi, tradotta e cercata in database proteina • tBLASTn: query proteina, tradotta all’indietro e cercata in database nucleotidi • tBLASTx: query nucleotidi, tradotta e cercata in database nucleotidi tradotti • megaBLAST: query nucleotidi vs genomi