Download

1 / 30
300 likes | 453 Views
Bioinformatics Pipeline for Fosmid based Molecular Haplotype Sequencing. Jorge Duitama 1,2 , Thomas Huebsch 1 , Gayle McEwen 1 , Sabrina Schulz 1 , Eun -Kyung Suk 1 , Margret R. Hoehe 1 1 . Max Planck Institute for Molecular Genetics, Berlin, Germany

E N D
Bioinformatics Pipeline for Fosmid based Molecular Haplotype Sequencing • Jorge Duitama1,2, Thomas Huebsch1, Gayle McEwen1, Sabrina Schulz1, Eun-Kyung Suk1, Margret R. Hoehe1 • 1. Max Planck Institute for Molecular Genetics, Berlin, Germany • 2. Department of Computer Science and Engineering University of Connecticut, Storrs, CT, USA
29,74 31,59 32,34 33,21 MHC class I MHC class III MHC class II MHC: Key Region for Common Diseases & Transplant Medicine
MHC: Variation amongst Haplotypes Variation of MHC Haplotypes against PGF reference RCCX CNV HLA-DRB CNV Variation and annotation map for eight MHC haplotypes, Horton et al. Immunogenetics (2008) 60,1-18 7 further MHC Haplotype sequences PGF reference sequence MHC class III MHC class II • Variation amongst 8 MHC Haplotypes: • 37.451 Substitutions • 7.093 Short Indels
40 kb haploid molecules 5000 fosmids 100 Individuals 100 Libraries One pool SNP Mapping for Prioritization of MHC Informative Pools Complete Fosmid Pool 3x96-well = 288 fosmid pools Contiguous MHC haplotype sequence Identification of 40 kb fosmid sequences Targeted Enrichment Haplotype A Haplotype B Phasing molecular fosmid sequences Experimental Approach SOLiD NGS Platform Shotgunning complete 40 kb fosmids Data Analysis Pipeline T G A
Data Analysis Pipeline Fosmid Detection Program Read Alignment against Genome Fosmid Specific Matching Algorithm Pairing Fosmid Sequences Based Phasing Visualization & MHC Database Consensus CallingSNP Analysis In House Project Specific Analysis Pipeline SOLiD Standard Pipeline
Data Analysis Pipeline Fosmid Detection Program Read Alignment against Genome Fosmid Specific Matching Algorithm Pairing Fosmid Sequences Based Phasing Consensus CallingSNP Analysis Visualization & MHC Database In House Project Specific Analysis Pipeline SOLiD Standard Pipeline
Mapping real data Pool of 15.000 Fosmids 22 Mill. Reads 50bp
Data Analysis Pipeline Fosmid Detection Program Read Alignment against Genome Fosmid Specific Matching Algorithm Pairing Fosmid Sequences Based Phasing Consensus CallingSNP Analysis Visualization & MHC Database In House Project Specific Analysis Pipeline SOLiD Standard Pipeline
SNP Calling Accuracy in the MHC • Affymetrix genotype information for 1583 SNP positions as reference standard: • - Homozygous identical with reference: 957 • - Heterozygous: 562 • - Homozygous different from reference: 64 • Compared to variants called from the SOLiD sequenced genomic DNA sample (15x average read coverage) • Percentage of error in genotype calling: 3.66% • False positive rate: 0.1% • False negative rate: 9.25%
Data Analysis Pipeline Fosmid Detection Program Read Alignment against Genome Fosmid Specific Matching Algorithm Pairing Fosmid Sequences Based Phasing Consensus CallingSNP Analysis Visualization & MHC Database In House Project Specific Analysis Pipeline SOLiD Standard Pipeline
Fosmids Detection • Fosmid Detection Algorithm • Assign each read to a single 1kb long bin. Select bins with more than 5 reads • Perform allele calls for each heterozygous SNP. Mark bins with heterozygous calls • Cluster adjacent bins as belonging to the same fosmid if: • The gap distance between them is less than 10kb and • There are no bins with heterozygous SNPs between them • Keep fosmids with lengths between 3kb and 60kb UCSC Genome browser http://genome.ucsc.edu/Kent et al. 2002 Genome Res.12(6):996-1006.
Fosmids Detection Size distribution of read-contigs 20 – 50 kb fosmid sized contigs
Data Analysis Pipeline Fosmid Detection Program Read Alignment against Genome Fosmid Specific Matching Algorithm Pairing Fosmid Sequences Based Phasing Consensus CallingSNP Analysis Visualization & MHC Database In House Project Specific Analysis Pipeline SOLiD Standard Pipeline
Haplotyping The process of grouping alleles that are present together on the same chromosome copy of an individual is called haplotyping
Single Individual Haplotyping • Input: Matrix M of m fragments covering n loci
Single Individual Haplotyping • Input: Matrix M of m fragments covering n loci
Single Individual Haplotyping • Input: Matrix M of m fragments covering n loci
Single Individual Haplotyping • Input: Matrix M of m fragments covering n loci
ReFHap Problem Formulation For two alleles a1, a2 For two rows i1, i2 of M s(M,1,2) = 1
ReFHap Problem Formulation For a cut I of rows of M
ReFHap Algorithm • Reduce the problem to Max-Cut. • Solve Max-Cut • Build haplotypes according with the cut 4 1 -1 3 1 2 1 -1 3 h1 00110 h2 11001
ReFHap Algorithm • Build G=(V,E,w) from M • Sort E from largest to smallest weight • Init I with a random subset of V • For each e in the first k edges • I’ ← GreedyInit(G,e) • I’ ← GreedyImprovement(G,I’) • If s(M, I) < s(M, I’) then I ← I’
1 4 1 4 2 3 2 3 ReFHap Algorithm • Classical greedy algorithm
1 2 3 4 ReFHap Algorithm • Edge flipping 2 1 3 4
Phasing MHC:Preliminary Results • Number of blocks: 8 • N50 block length: 793 kb • Maximum block length: 1.6 MB • Total extent of all blocks: 3.8 MB • Fraction of MHC phased into haplotype blocks: 95% • Number of heterozygous SNPs: 8030 SNPs • Fraction of SNPs phased: 86%
Acknowledgements Anita Suk Thomas Hübsch Margret Hoehe Roger Horton Gayle McEwen Steffi Palczewski Britta Horstmann Sabrina Schulz The Life Tech Team: Kevin McKernan Alexander Sartori Clarence Lee Dustin Holloway Jessica Spangler Heather Peckham Tristen Weaver Stephen McLaughlin Tamara Gilbert Tim Harkins Thank You!
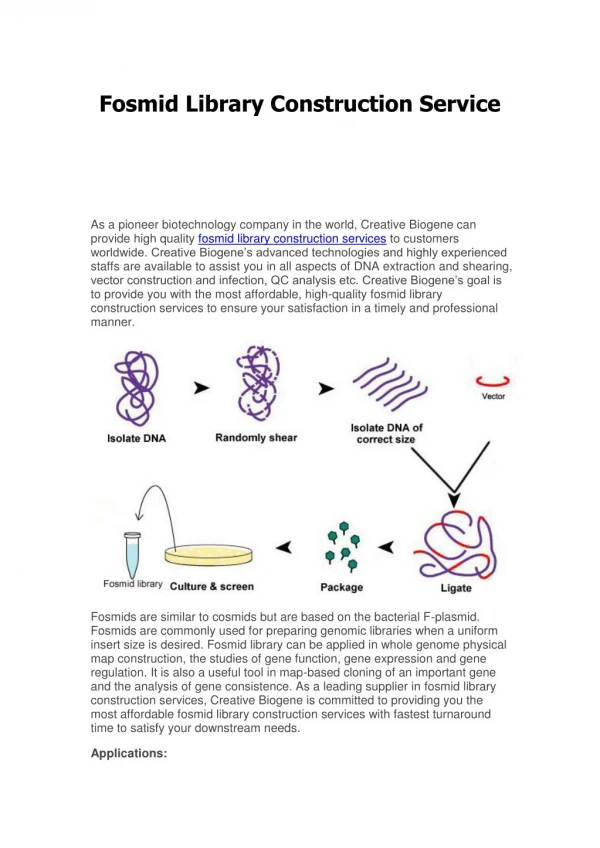
Comparison Mapping algos COX Haplotype simulated reads