Download

1 / 61
610 likes | 717 Views
3D Structures of Biological Macromolecules Part 5: Protein Structure Prediction and Protein Folding. Jürgen Sühnel jsuehnel@fli-leibniz.de. Leibniz Institute for Age Research, Fritz Lipmann Institute, Jena Centre for Bioinformatics Jena / Germany.

E N D
3D Structures of Biological Macromolecules Part 5: Protein Structure Prediction and Protein Folding Jürgen Sühnel jsuehnel@fli-leibniz.de Leibniz Institute for Age Research, Fritz Lipmann Institute, Jena Centre for Bioinformatics Jena / Germany Supplementary Material: http://www.fli-leibniz.de/www_bioc/3D/
Molecular Mechanics (Force Field) http://cmm.info.nih.gov/modeling/guide_documents/molecular_mechanics_document.html
How Do We Get the Parameters ? Experimental Data (Examples: Geometrical Parameters) Quantum-chemical Calculations (Examples: Charges)
Optimization Methods – Steepest Descent Selection of an initial point x0 Determination of direction and step size for calculating the next point
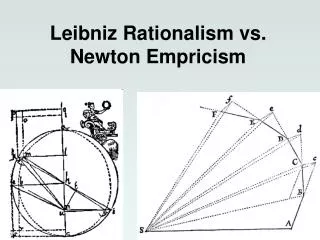
Optimization Methods – Newton-Raphson Methods g -. gradient h - Hessian
Simulation of Protein Folding – Molecular Dynamics AMBER GROMOS CHARMM TINKER
1HGV, extended structure 1HGV, actual structure 1HGV, 61% helix, 1.928 ns 1HGV, 75% helix, 3.428 ns Molecular Dynamics Simulation Protein Capsid Of Filamentous Bacteriophage Ph75 From Thermus Thermophilus Images created using VMD (Visual Molecular Dynamics) (HUMPHREY, W., DALKE, A. and SCHULTEN, K., 1996.VMD - Visual Molecular Dynamics. Journal Molecular Graphics,14, pp33-38).
Molecular Dynamics Packages amber.scripps.edu
Molecular Dynamics Packages www.igc.ethz.ch/gromos/
Molecular Dynamics Packages www.charmm.org
Molecular Dynamics Packages dasher.wustl.edu/tinker/
Visualizing and Analyzing Molecular Dynamics Simulations www.ks.uiuc.edu/Research/vmd/
Folding Surface for Lysozyme Dobson, Sali, Karplus, Angew. Chem. Int. Ed.1998, 37, 868.
Protein Folding States Dobson, Sali, Karplus, Angew. Chem. Int. Ed.1998, 37, 868.
Monitoring Protein Folding by Experimental Methods Dobson, Sali, Karplus, Angew. Chem. Int. Ed.1998, 37, 868.
Monitoring Protein Folding by Experimental Methods Paxco, Dobson, Curr. Opin. Struct. Biol.1996, 6, 630.
Protein Folding by Molecular Dynamics Villin headpiece domain (PDB code: 1vii) Actin binding site highlighted 36 amino acids
Radius of Gyration The radius of gyration Rgis defined by the root-mean-square distance between all atoms in a molecule and the centroid. In a globular protein the radius of gyration Rg can be predicted with reasonable accuracy from the relationship Rg(pred) = 2.2 N 0.38 where N is the number of amino acids.
Statistical Potentials wij(r) – interaction free energy ij(r) - pair density * - reference pair density at infinite separation Statistical potentials can be determined by simply counting interactions of a specific type in a dataset of experimental structures. The distance dependence may or may not be taken into account. If not, the interaction free energy is usuallycalled a contact potential. It represents an average over distances shorter than some cutoff distance rc. Thomas, Dill, J. Mol. Biol.1996, 257, 457-469
Lattice Algorithm • Red = hydrophobic, Blue = hydrophilic • If Red is near empty space E = E+1 • If Blue is near empty space E = E-1 • If Red is near another Red E = E-1 • If Blue is near another Blue E = E+0 • If Blue is near Red E = E+0
Ab Initio Protein Structure Prediction http://rosettadesign.med.unc.edu/
Ab Inition Protein Structure Prediction - Rosetta Structure representation: Only main-chain heavy atoms and Cbeta-atom of sidechains are taken into account, Bond lengths and bond angles are held constant and correspond to the alanine geometry. The only remaining geometrical variables are the backbone torsion angles. Structure generation: Generation of fragment libraries from experimental structures (3 and 9 amino acids). Splicing together fragments of proteins of known structure with similar sequences. The conformational space defined by these fragments is then searched by a Monte Carlo procedure with an energy function that favors compact structures with paired beta-strands and buried hydrophobic amino acids. A total of 1000 independent simulations are carried out (starting from different random number seeds) for each query sequence. The resulting structures are clustered. Initial evaluation by a scoring function Low-scoring conformations are identified by simulated annealing with a move set that involves replacing the torsion angles of a segment of the chain with a related amino acid sequence. Further evaluation.
ROSETTA Results Simons, Strauss, Baker. J. Mol. Biol.2001, 306, 1191-1199.
Computational Thermostabilization PDB code: 1ox7 Cytosine deaminase (CD) catalyzes the deamination of cytosine (converts cytosine to uracil) and is only present in prokaryotes and fungi, where it is a member of the pyrimidine salvage pathway. The enzyme is of interest both for antimicrobial drug design and gene therapy applications against tumors.
Computational Thermostabilization Prediction of stabile mutations with Rosetta Design