Download

1 / 20
200 likes | 302 Views
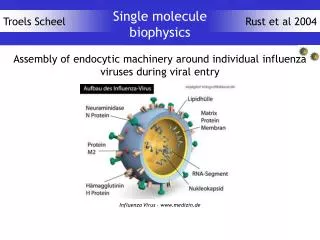
Sequence Assembly for Single Molecule Methods. Steven Skiena, Alexey Smirnov Department of Computer Science SUNY at Stony Brook {skiena, alexey}@cs.sunysb.edu. The State of Sequence Assembly.

E N D
Sequence Assembly for Single Molecule Methods Steven Skiena, Alexey Smirnov Department of Computer Science SUNY at Stony Brook {skiena, alexey}@cs.sunysb.edu
The State of Sequence Assembly The success of full genome sequencing implies that shotgun sequence assembly with current technologies is largely a solved problem With conventional sequence technologies: • read length: about 500 base pairs • error rate: under 2% • coverage: about 10 times for bacteria, about 30 times for humans But single molecule sequencing methods promise to change these parameters significantly
Single Molecule Sequencing Methods Single molecule sequencing methods, such as being developed byU.S.Genomics, promise much longer readlengths • read length: hundreds of thousands of bases ? • error rate: ? "No free lunch hypothesis"- we anticipate that the new technologies will (at least initially) have significantly higher error rates than current sequencing machines. Our assumption – long lousy reads.
Our Problems • What levels of coverage will be needed to get accurate sequence informationfrom long noisy reads? • How do we efficiently assemble such long noisy reads?
Sequencing from Subsequences Why subsequences? • We anticipate that certain single moleculesequencingtechnologies will be prone tohaving manybase deletion errors • Example: in the U.S. Genomicstechnology, sequence bases are replacedby tagged bases.Untagged bases are invisible, generatingsubsequences. We study the effect of per base deletion frequencies on our ability to accuratelyreconstruct long sequences.Our study revolves around this theoretical error model. But our algorithm can be easily generalized.
Notation • n – length of the original sequence; • p – base deletion rate; • k – number of reads; • Ri – a read of the original sequence;
Quality of Reconstruction Metric Our score function is • where ED is the edit distance, s is the target sequence of length n, s’ – sequence reconstructed from the reads. • An empty string has a score of 0; • The target string has a score of 1;
Lower Bounds on Reconstruction Quality • k=0 -> report a random string of some length. Computational experiments showed that reporting a string of length 0.6n gives best results (score=0.37) • k=1 -> report this read; score=1-p (because (1-p)*n characters will be matched and the rest will be inserted).
Information Theory Bounds What is the minimal number of reads that we need to reconstruct the sequence? First, we need to know the number of sequences of length n in which a given read of length k occurs: Each of reads gives us at most this number of bits of information: Therefore, we will need at least this many reads:
Bounds on the Number of Reads Conclusion: reconstruction becomes impossible for error rates higher than 75%, but possible for 50%
Sequence Assembly Algorithm We use a two phase procedure: • Insertion: align a read Ri with consensus sequence Ci-2 and build a new consensus Ci-1 • Refining and Cleanup: delete/reorder characters from current consensus to better reflect the reads and delete unused characters refine & cleanup C3 C2 R4 refine & cleanup C1 refine & cleanup R3 R1 R2
Read Insertion How to choose the optimal alignment to insert a new read into current consensus Ci? Pairwise align all reads against Ci and for each position of Ci, compute the number of times each particular character was inserted into it at this position. Align the read being inserted against the weighted consensus sequence using the insertion weights generated before.
Consensus Refining Pairwise alignment from reads is prone to two types of errors: inserting a pair of characters in a wrong order and undersampling R1 : ATA refine ATCA ACTAA ACTAA R2 : ACA Solution: Try to make a swap and a character doubling at each position and see if it improves the alignment score for some reads.
Clean up Procedure • Pairwise align all reads against the target to weight the positions of S by frequency of use. • Update weights after each alignment to bias matches toward frequently used positions. • Delete all characters matched fewer than a certain number of times.
Complexity Analysis • Each insertion step takes O(k*n*n) time • Each refining step takes O(k*n*n) time • Each cleanup step takes O(k*n*n) time • Total: O(niter*k*n*n) where niter is the number of iterations
Results • For base deletion rates as high as 40%, we can completely reconstruct sequences with high enough coverage (50 times coverage) • For larger error rates, our algorithm finds shorter supersequences, i.e. there are multiple answers so exact reconstruction is impossible. • Here we ignored the possibility of insertion/substitution errors, but it is clear our methods can adapt to different error models at lower error rates.
Future Work • We want to build your single molecule sequence assembler! • Our Stroll shotgun sequence assembler (Chen and Skiena) was used by Brookhaven National Laboratory to sequence the bacterial Borrelia burgdorferi. • We are particularly interested in identifying better error models for sequencing technologies under current development.
The End Questions?