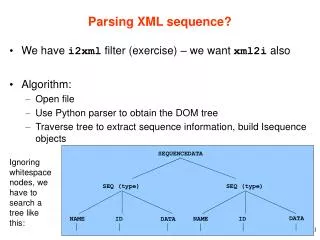
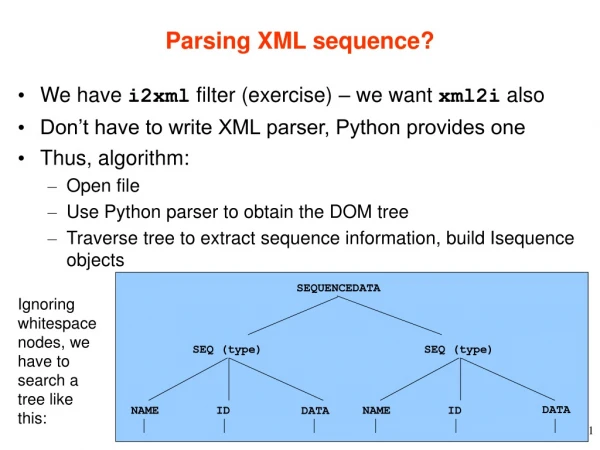
Download
1 / 17
180 likes | 344 Views
Sequence File Parsing using Biopython. BCHB524 2012 Lecture 10. Review. Modules in the standard-python library: sys, os, os.path – access files, program environment zipfile, gzip – access compressed files directly urllib – access web-resources (URLs) as files

E N D
Sequence File Parsing using Biopython BCHB5242012Lecture 10 BCHB524 - 2012 - Edwards
Review • Modules in the standard-python library: • sys, os, os.path – access files, program environment • zipfile, gzip – access compressed files directly • urllib – access web-resources (URLs) as files • csv – read delimited line based records from files • Plus lots, lots more. BCHB524 - 2012 - Edwards
BioPython • Additional modules that make many common bioinformatics tasks easier • File parsing (many formats) & web-retrieval • Formal biological alphabets, codon tables, etc • Lots of other stuff… • Have to install separately • Not part of standard python, or Enthought • biopython.org BCHB524 - 2012 - Edwards
Biopython: Fasta format • Most common biological sequence data format • Header/Description line • >accession description • Multi-accession sometimes represented • accession1|accession2|accession3 • lots of variations, no standardization • No prescribed format for the description • Other lines • sequence, one chunk per line. • Usually all lines, except the last, are the same length. BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO import Bio.SeqIOimport sys# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the FASTA file and iterate through its sequencesseqfile = open(seqfilename)for seq_record in Bio.SeqIO.parse(seqfile, "fasta"):# Print out the various elements of the SeqRecordprint"\n------NEW SEQRECORD------\n"print"seq_record.id:\n\t", seq_record.idprint"seq_record.description:\n\t",seq_record.descriptionprint"seq_record.seq:\n\t",seq_record.seqseqfile.close() BCHB524 - 2012 - Edwards
Biopython: Other formats • Genbank format • From NCBI, also format for RefSeq sequence • UniProt/SwissProtflat-file format • From UniProt for SwissProt and TrEMBL • UniProt-XML format: • From UniProt for SwissProtand TrEMBL • Use the gzip module to handle compressed sequence databases BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO import Bio.SeqIOimport sys# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the FASTA file and iterate through its sequencesseqfile = open(seqfilename)for seq_record in Bio.SeqIO.parse(seqfile, "genbank"):# Print out the various elements of the SeqRecordprint"\n------NEW SEQRECORD------\n"print"seq_record.id:\n\t", seq_record.idprint"seq_record.description:\n\t",seq_record.descriptionprint"seq_record.seq:\n\t",seq_record.seqseqfile.close() BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO import Bio.SeqIOimport sys# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the FASTA file and iterate through its sequencesseqfile = open(seqfilename)for seq_record in Bio.SeqIO.parse(seqfile, "swiss"):# Print out the various elements of the SeqRecordprint"\n------NEW SEQRECORD------\n"print"seq_record.id:\n\t", seq_record.idprint"seq_record.description:\n\t",seq_record.descriptionprint"seq_record.seq:\n\t",seq_record.seqseqfile.close() BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO import Bio.SeqIOimport sys# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the FASTA file and iterate through its sequencesseqfile = open(seqfilename)for seq_record in Bio.SeqIO.parse(seqfile, "uniprot-xml"):# Print out the various elements of the SeqRecordprint"\n------NEW SEQRECORD------\n"print"seq_record.id:\n\t", seq_record.idprint"seq_record.description:\n\t",seq_record.descriptionprint"seq_record.seq:\n\t",seq_record.seqseqfile.close() BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO and gzip import Bio.SeqIOimport sysimport gzip# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the FASTA file and iterate through its sequencesseqfile = gzip.open(seqfilename)for seq_record in Bio.SeqIO.parse(seqfile, "fasta"):# Print out the various elements of the SeqRecordprint"\n------NEW SEQRECORD------\n"print"seq_record.id:\n\t", seq_record.idprint"seq_record.description:\n\t",seq_record.descriptionprint"seq_record.seq:\n\t",seq_record.seqseqfile.close() BCHB524 - 2012 - Edwards
What about the other "stuff" • BioPython makes it easy to get access to non-sequence information stored in "rich" sequence databases • Annotations • Cross-References • Sequence Features • Literature BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO import Bio.SeqIOimport sysimport gzip# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the FASTA file and iterate through its sequencesseqfile = gzip.open(seqfilename)for seq_record in Bio.SeqIO.parse(seqfile, "uniprot-xml"):# What else is available in the SeqRecord?print"\n------NEW SEQRECORD------\n"print"repr(seq_record)\n\t",repr(seq_record)print"dir(seq_record)\n\t",dir(seq_record)breakseqfile.close() BCHB524 - 2012 - Edwards
BioPython: Bio.SeqRecord import Bio.SeqIOimport sysimport gzip# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the FASTA file and iterate through its sequencesseqfile = gzip.open(seqfilename)for seq_record in Bio.SeqIO.parse(seqfile, "uniprot-xml"):# Print out the various elements of the SeqRecordprint"\n------NEW SEQRECORD------\n"print"seq_record.annotations\n\t",seq_record.annotationsprint"seq_record.features\n\t",seq_record.featuresprint"seq_record.dbxrefs\n\t",seq_record.dbxrefsprint"seq_record.format('fasta')\n",seq_record.format('fasta')breakseqfile.close() BCHB524 - 2012 - Edwards
BioPython: Random access • Sometimes you want to access the sequence records "randomly"… • …to pick out the ones you want (by accession) • Why not make a dictionary, with accessions as keys, and SeqRecord values? • Use SeqIO.to_dict(…) • What if you don't want to hold it all in memory • Use SeqIO.index(…) BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO.to_dict(…) import Bio.SeqIOimport sys# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Open the sequence databaseseqfile = open(seqfilename)# Use to_dict to make a dictionary of sequence recordssprot_dict = Bio.SeqIO.to_dict(Bio.SeqIO.parse(seqfile, "uniprot-xml"))# Close the fileseqfile.close()# Access and print a sequence recordprint sprot_dict['Q6GZV8'] BCHB524 - 2012 - Edwards
BioPython: Bio.SeqIO.index(…) import Bio.SeqIOimport sys# Check the inputiflen(sys.argv) < 2:print >>sys.stderr, "Please provide a sequence file" sys.exit(1)# Get the sequence filenameseqfilename = sys.argv[1]# Use to_dict to make a dictionary of sequence recordssprot_dict = Bio.SeqIO.index(seqfilename, "uniprot-xml")# Access and print a sequence recordprint sprot_dict['Q6GZV8'] BCHB524 - 2012 - Edwards
Exercises • Read through and try the examples from Chapters 2-5 of BioPython's Tutorial. • Download human proteins from RefSeq and compute amino-acid frequencies for the (RefSeq) human proteome. • Which amino-acid occurs the most? The least? • Hint: access RefSeq human proteins fromftp://ftp.ncbi.nih.gov/refseq • Download human proteins from SwissProt and compute amino-acid frequencies for the SwissProt human proteome. • Which amino-acid occurs the most? The least? • Hint: access SwissProt human proteins from http://www.uniprot.org/downloads -> “Taxonomic divisions” • How similar are the human amino-acid frequencies of in RefSeq and SwissProt? BCHB524 - 2012 - Edwards