Download

1 / 31
380 likes | 815 Views
Thalessemia. Pathogenesis. Genetically determined Heterogenous group of disorder Reduced synthesis of one or more types of normal haemoglobin polypeptide chains. Demographics: Thalassemia. Found most frequently in the Mediterranean, Africa, Western and Southeast Asia, India and Burma.

E N D
Pathogenesis • Genetically determined • Heterogenous group of disorder • Reduced synthesis of one or more types of normal haemoglobin polypeptide chains
Demographics: Thalassemia • Found most frequently in the Mediterranean, Africa, Western and Southeast Asia, India and Burma
Normal Haemoglobin • HbA - α2β2 • HbA2 - α2δ2 • HbF – α2γ2
Each goblin chain have separate genetic control α –thalassaemia affect α-chain synthesis β –thalassaemia affect β-chain synthesis
β-Thalassaemia β Chain synthesis An absence or deficiency of β-chain synthesis of adult HbA Hb-A γ and δ chain Hb-A = α2β2
Pathophysiology of β-Thalassaemia mutation in β-gene Complete or partial absence of β-chain Decreased adult HbA α-chain synthesis remain normal Free complementary α-chain – unstable and precipitate within normoblasts as insoluble inclusions Cell membrane damage & impaired DNA synthesis apoptosis i.e. ineffective erythropoeisis
On the basis of synthetic ability β-genes are designated as • β gene – can synthesize normal amount of β-chain • β+ gene – can synthesize reduced amount of β-chain • β0 gene – cannot synthesize β-chain
β-thalassaemia major • Mutation of normal β-gene β0-gene absence HbA increased HA2 and HbF • genotype – β0β0 • β-thalassaemia intermedia • ↑HbA2 • ↑HbF • ↓HbA • Genotype β+β+ or β0β • β-thalassaemia minor • ↑HbA2 • HbA normal • HbF normal
Symptoms • Inadequate production + ineffective erythropoiesis + haemolysis Anaemia • hemolysis hyperbillirubinemia jaundice • To compensate anaemia extramedullary haemopoiesis in liver,spleen Hepatosplenomegaly • ↑Erythropoiesis marrow expansion & thinning of cortex of skull bone Thalassaemia facies/bone pain • Iron overload—increased absorption and transfusions Endocrine disorders, Cardiomyopathy, Liver failure
management • Regular blood transfusions • Iron chelation-desferroxamine SC infusion/oral • Splenectomy • Monitor for complications of iron overload-LFT,ECHO,Thyroid functions,growth (pituitary can be damaged)
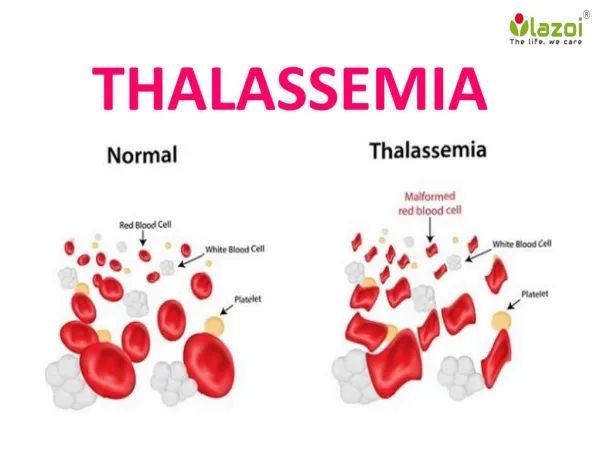
Normal Thalassaemia
α-Thassaemia An absence or deficiency of α-chain synthesis due to delation of α-genes.
Pathogenesis of α-Thalassaemia • In normal individual HbA, HbA2 and HbF need α-chain for their formation. • 4 genes of α-chain, each pair on short arm of chromosome 16 • In α-thalassaemia, delation of α-genes reduction or absence of synthesis of α-chain depending on number of α-gene deletion.
↓α-chain synthesis free γ-chain in the fetus & β-chain in infant of 6 months, and continue in the rest of life. • Complementary 4γ and 4β are aggregated Hb Bart (4γ ) and HbH (4β ), respectively.
Variants of α-Thalassaemia • Silent carrier • Delation of single α-gene • Genotype α/αα • Asymptomatic • Absence of RBC abnormality • Thalasaemia trait • Delation of 2 α-genes • Genotype --/αα • Asymptometic, minimal or no anaemia • Minimal RBC abnormalities
Hb H disease • Delation of 3 α-genes • Genotype --/- α • 75% reduction of α-chain • small amount of HbF, HbA, & HbA2 • Fetus can survive • Severe anaemia • Hydrops fetalis • Delation of all α-genes • Genotype --/-- • Absence of α-chain synthsis • Only Hb Bart (γ4) is produced • Incompatible with life
Inheritance • Autosomal recessive • Inc incidence in consanguineous marriages • Can be prevented by detecting carriers
Laboratory Findings • Hb – low • RBC count – Markedly decreased • MCV, MCH, MCHC – reduced • Blood picture-abn RBC • Hb electrophoresis –can demonstrate the types of Hb present
Peripheral Blood Film Normal