Download
1 / 1
10 likes | 164 Views
SPECIFIC INHIBITION OF THE REDOX ACTIVITY OF THE MULTIFUNCTIONAL APE1/REF-1 PROTEIN BY E3330 BLOCKS TNF- a -INDUCED ACTIVATION OF IL-8 PRODUCTION IN HEPATIC CELL LINES E. Codarin 1 , L. Cesaratto 1,2 , A. Caragnano 1 , S. Bellentani 2,3 *, M.R. Kelley 4 , C. Tiribelli 2 , G. Tell 1 .

E N D
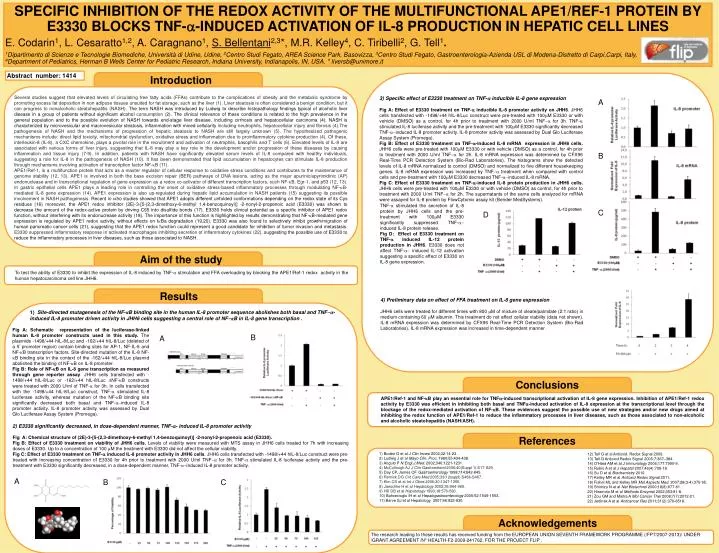
SPECIFIC INHIBITION OF THE REDOX ACTIVITY OF THE MULTIFUNCTIONAL APE1/REF-1 PROTEIN BY E3330 BLOCKS TNF-a-INDUCED ACTIVATION OF IL-8 PRODUCTION IN HEPATIC CELL LINES E. Codarin1, L. Cesaratto1,2, A. Caragnano1, S. Bellentani2,3*, M.R. Kelley4, C. Tiribelli2, G. Tell1. 1Dipartimento di Scienze e Tecnologie Biomediche, Università di Udine, Udine, 2Centro Studi Fegato, AREA Science Park, Basovizza, 3Centro Studi Fegato, Gastroenterologia-Azienda USL di Modena-Distretto di Carpi,Carpi, Italy, 4Department of Pediatrics, Herman B Wells Center for Pediatric Research, Indiana University, Indianapolis, IN, USA. * liversb@unimore.it Abstract number: 1414 Introduction Several studies suggest that elevated levels of circulating free fatty acids (FFAs) contribute to the complications of obesity and the metabolic syndrome by promoting excess fat deposition in non adipose tissues unsuited for fat storage, such as the liver (1). Liver steatosis is often considered a benign condition, but it can progress to nonalcoholic steatohepatitis (NASH). The term NASH was introduced by Ludwig to describe histopathology findings typical of alcoholic liver disease in a group of patients without significant alcohol consumption (2). The clinical relevance of these conditions is related to the high prevalence in the general population and to the possible evolution of NASH towards end-stage liver disease, including cirrhosis and hepatocellular carcinoma (4). NASH is characterized by microvesicular and macrovesicular steatosis, inflammation with mixed cellularity including neutrophils, hepatocellular injury and fibrosis (4).The pathogenesis of NASH and the mechanisms of progression of hepatic steatosis to NASH are still largely unknown (5). The hypothesized pathogenic mechanisms include: direct lipid toxicity, mitochondrial dysfunction, oxidative stress and inflammation due to proinflammatory cytokine production (4). Of these, interleukin-8 (IL-8), a CXC chemokine, plays a pivotal role in the recruitment and activation of neutrophils, basophils and T cells (6). Elevated levels of IL-8 are associated with various forms of liver injury, suggesting that IL-8 may play a key role in the development and/or progression of these diseases by causing inflammation and tissue injury (7-9). In addition, patients with NASH have significantly elevated serum levels of IL-8 compared with healthy individuals, suggesting a role for IL-8 in the pathogenesis of NASH (10). It has been demonstrated that lipid accumulation in hepatocytes can stimulate IL-8 production through mechanisms involving activation of transcription factor NF-kB (11). APE1/Ref-1, is a multifunction protein that acts as a master regulator of cellular response to oxidative stress conditions and contributes to the maintenance of genome stability (12, 13). APE1 is involved in both the base excision repair (BER) pathways of DNA lesions, acting as the major apurinic/apyrimidinic (AP) endonuclease and in transcriptional regulation of gene expression as a redox co-activator of different transcription factors, such NF-kB, Egr-1, and p53 (12, 13). In gastric epithelial cells APE1 plays a leading role in controlling the onset of oxidative stress-based inflammatory processes through modulating NF-kB-mediated IL-8 gene expression (14). APE1 expression is also up-regulated during hepatic lipid accumulation in NASH patients (15) suggesting its possible involvement in NASH pathogenesis. Recent in vitro studies showed that APE1 adopts different unfolded conformations depending on the redox state of its Cys residues (16) moreover, the APE1 redox inhibitor (2E)-3-[5-(2,3-dimethoxy-6-methyl 1,4-benzoquinoyl)] -2-nonyl-2-propenoic acid (E3330) was shown to decrease the amount of the redox-active protein by driving C65 into disulfide bonds (17). E3330 holds clinical potential as a specific inhibitor of APE1 redox function, without interfering with its endonuclease activity (18). The importance of this function is highlighted by results demonstrating that NF-kB-mediated gene expression is regulated by APE1 redox activity, without effects on IkBa degradation (19,20). E3330 was also found to selectively inhibit growth/migration of human pancreatic cancer cells (21), suggesting that the APE1 redox function could represent a good candidate for inhibition of tumor invasion and metastasis. E3330 suppressed inflammatory response in activated macrophages inhibiting secretion of inflammatory cytokines (22), suggesting the possible use of E3330 to reduce the inflammatory processes in liver diseases, such as those associated to NASH. 3) Specific effect of E3330 treatment on TNF-a inducible IL-8 gene expression Fig A: Effect of E3330 treatment on TNF-a inducible IL-8 promoter activity on JHH6. JHH6 cells transfected with -1498/+44 hIL-8/Luc construct were pre-treated with 100mM E3330 or with vehicle (DMSO) as a control, for 4h prior to treatment with 2000 U/ml TNF-a for 3h. TNF-a stimulated IL-8 luciferase activity and the pre-treatment with 100mM E3330 significantly decreased TNF-a-induced IL-8 promoter activity. IL-8 promoter activity was assessed by Dual Glo Luciferase Assay System (Promega). Fig B: Effect of E3330 treatment on TNF-a-induced IL-8 mRNA expression in JHH6 cells. JHH6 cells were pre-treated with 100mM E3330 or with vehicle (DMSO) as a control, for 4h prior to treatment with 2000 U/ml TNF-a for 2h. IL-8 mRNA expression was determined by CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories). The histograms show the detected levels of IL-8 mRNA normalized to control (DMSO) and normalized to five different housekeeping genes. IL-8 mRNA expression was increased by TNF-a treatment when compared with control cells and pre-treatment with 100mM E3330 decreased TNF-a-induced IL-8 mRNA. Fig C: Effect of E3330 treatment on TNF-a-induced IL-8 protein production in JHH6 cells. JHH6 cells were pre-treated with 100mM E3330 or with vehicle (DMSO) as control, for 4h prior to treatment with 2000 U/ml TNF-a for 2h. The supernatants of the same cells analyzed for mRNA were assayed for IL-8 protein by FlowCytomix assay kit (Bender MedSystems). A B TNF-a stimulated the secretion of IL-8 protein by JHH6 cells and the pre-treatment with 100mM E3330 significantly suppressed TNF-a- induced IL-8 protein release. Fig D: Effect of E3330 treatment on TNF-a induced IL-12 protein production in JHH6. E3330 does not affect TNF-a- induced IL-12 activation suggesting a specific effect of E3330 on IL-8 gene expression. C D Aim of the study To test the ability of E3330 to inhibit the expression of IL-8 induced by TNF-a stimulation and FFA overloading by blocking the APE1/Ref-1 redox activity in the human hepatocarcinoma cell line JHH6. Results 4) Preliminary data on effect of FFA treatment on IL-8 gene expression JHH6 cells were treated for different times with 800 mM of mixture of oleate/palmitate (2:1 ratio) in medium containing 60 mM albumin. This treatment do not affect cellular viability (data not shown). IL-8 mRNA expression was determined by CFX96 Real-Time PCR Detection System (Bio-Rad Laboratories). IL-8 mRNA expression was increased in time-dependent manner . 1) Site-directed mutagenesis of the NF-kB binding site in the human IL-8 promoter sequence abolishes both basal and TNF-a-induced IL-8 promoter driven activity in JHH6 cells suggesting a central role of NF-kB in IL-8 gene transcription . Fig A: Schematic representation of the luciferase-linked human IL-8 promoter constructs used in this study. The plasmids -1498/+44 hIL-8/Luc and -162/+44 hIL-8/Luc (deleted of a 5’ promoter region) contain binding sites for AP-1, NF-IL-6 and NF-kB transcription factors. Site-directed mutation of the IL-8 NF-kB binding site in the context of the -162/+44 hIL-8/Luc plasmid abolished the binding of NF-kB on IL-8 promoter. Fig B: Role of NF-kBon IL-8 gene transcription as measured through gene reporter assay. JHH6 cells transfected with -1498/+44 hIL-8/Luc or -162/+44 hIL-8/Luc DNF-kB constructs were treated with 2000 U/ml of TNF-a for 3h. In cells transfected with the -1498/+44 hIL-8/Luc construct, TNF-a stimulated IL-8 luciferase activity, whereas mutation of the NF-kB binding site significantly decreased both basal and TNF-a-induced IL-8 promoter activity. IL-8 promoter activity was assessed by Dual Glo Luciferase Assay System (Promega). B A Conclusions APE1/Ref-1 and NF-kB play an essential role for TNFa-induced transcriptional activation of IL-8 gene expression. Inhibition of APE1/Ref-1 redox activity by E3330 was efficient in inhibiting both basal and TNFa-induced activation of IL-8 expression at the transcriptional level through the blockage of the redox-mediated activation of NF-kB. These evidences suggest the possible use of new strategies and/or new drugs aimed at inhibiting the redox function of APE1/Ref-1 to reduce the inflammatory processes in liver diseases, such as those associated to non-alcoholic and alcoholic steatohepatitis (NASH/ASH). 2) E3330 significantlydecreased, in dose-dependentmanner, TNF-a-induced IL-8 promoter activity Fig A: Chemical structure of (2E)-3-[5-(2,3-dimethoxy-6-methyl 1,4-benzoquinoyl)] -2-nonyl-2-propenoic acid (E3330). Fig B: Effect of E3330 treatment on viability of JHH6 cells. Levels of viability were measured with MTS assay in JHH6 cells treated for 7h with increasing doses of E3330. Up to a concentration of 100 mM the treatment with E3330 did not affect the cellular viability. Fig C: Effect of E3330 treatment on TNF-a induced IL-8 promoter activity in JHH6 cells. JHH6 cells transfected with -1498/+44 hIL-8/Luc construct were pre-treated with increasing concentration of E3330 for 4h prior to treatment with 2000 U/ml TNF-a for 3h. TNF-a stimulated IL-8 luciferase activity and the pre-treatment with E3330 significantly decreased, in a dose-dependent manner, TNF-a-induced IL-8 promoter activity. References 1) Boden G et al J Clin Invest 2002;32:14-23. 2) Ludwig J et al Mayo Clin. Proc. 1980;55:434-438. 3) Angulo P N.Engl.J.Med. 2002;346:1221-1231. 4) McCullough AJ J Clin Gastroenterol 2006;40(Suppl 1):S17-S29. 5) Day CP, James OF Gastroenterology 1998;114:842-845. 6) Remick DG Crit Care Med 2005;33(12suppl):S466-S467. 7) Kim CS et al Int J Obes 2006;30:1347-1355. 8) Jaeschke H et al Hepatology 2002;35:964-965. 9) Hill DB et al Hepatology 1993;18:576-580. 10) Bahcecioglu IH et al Hepatogastroenterology 2005;52:1549-1553. 11) Barve SJ et al Hepatology 2007;46:823-830. 12) Tell G et al Antioxid. Redox Signal 2009. 13) Tell G Antioxid Redox Signal 2005;7:367–384. 14) O’Hara AM et al J Immunology 2006;177:7990-9. 15) Rubio A et al J Hepatol2007;46(4):708-18. 16) Su D et al Biochemistry 2010. 17) Kelley MR et al Antioxid Redox Signal2011. 18) Fishel ML and Kelley MR Mol Aspects Med. 2007;28(3-4):375-95. 19) Shimizu N et al Nat Biotechnol 2000;18(8):877-81. 20)Hiramoto M et al Methods Enzymol 2002;353:81-8. 21) Zou GM and Maitra A Mol Cancer Ther 2008;7(7):2012-21. 22) Jedinak A et al Anticancer Res 2011;31(2):379-8518. A B C Acknowledgements The research leading to these results has received funding from the EUROPEAN UNION SEVENTH FRAMEWORK PROGRAMME (/FP7/2007-2013)/ UNDER /GRANT AGREEMENT /N° HEALTH-F2-2009-241762, FOR THE PROJECT FLIP .