Download

1 / 86
860 likes | 871 Views
Discover the evolution of protein database search programs from BLAST 1.0 to PSI-BLAST, including gapped alignment, statistical preliminaries, and performance evaluation. Dive into the optimized algorithms, scoring systems, and heuristics that make BLAST a powerful tool for sequence similarities. Explore the two-hit method and gapped alignment as enhancements in BLAST 2.0, with insights on sensitivity, speed, and alignment strategies. Learn how to maximize search efficiency and accuracy in the new generation of protein database searches.

E N D
Gapped BLAST and PSI-BLAST:a new generation of protein database search programs Presented by 佘健生 鄭為正 李定達 曾文鴻
Outline • BLAST 1.0 • BLAST 2.0 • The two-hit method • Gapped alignment • PSI-BLAST • Performance evaluation • Discussion and Conclusion • NCBI website
Statistical preliminaries • HSP: High-scoring segment pair • Locally optimal pair • S’ = (λS - ㏑K) / ㏑2 • S’: normalized score • Pi : background probability that amino acids occur randomly at all position • sij: score for aligning each pair of amino acids I and j • K : minor constant • λ: constant to adjust for matrix • sij and Pi → K and λ
E = N / 2S’ • E: number of distinct HSPs with normalized score at least S’ • N = mn is search space • S’ = log2(N/E) • qij = PiPjeλuSij • qij : target frequency of aligned pair of letters (i, j) with HSP, high-scoring segment paris • λu: the unique positive number
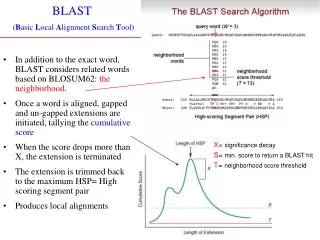
BLAST • Basic Local Alignment Search Tool(by Altschul, Gish, Miller, Myers and Lipman) • The BLAST program are widely used tools for searching protein and DNA database for sequence similarities • BLAST is a heuristic that attempts to optimize a specific similarity measure. • The central idea of the BLAST algorithm is that a statistically significant alignment is likely to contain a high-scoring pair of aligned words.
The maximal segment pair measure • MSP(maximal segment pair): the highest scoring pair of identical length segments chosen from 2 sequences • for DNA: Identities: +5; Mismatches: -4 • for protein: BLOSUM62 … • BLAST heuristically attempts to calculate the MSP score. • DP is O(mn) ,but BLAST is O(m) the highest scoring pair
BLAST 1.0 • Build the hash table for Sequence A. • Scan Sequence B for hits. • Extend hits.
For DNA : Seq. A = ACGTAGTA 12345678 AAA AAC .. ACG 1 .. AGT 5 .. CGT 2 .. GTA 3 6 .. TAG 4 .. TTT For protein : Seq. A = YGGFMAdd xyz to the hash table if Score(xyz, YGG) ≧ T;Add xyz to the hash table if Score(xyz, GGF) ≧ T;Add xyz to the hash table if Score(xyz, GFM) ≧ T; T: ‘threshold’ parameter High T yelds greater speed, but weak similarities Step 1: Build the hash table for Sequence A. (3-tuple example) Hash table
List all words in query YGGFMTSEKSQTPLVTLFKNAIIKNAHKKGQ YGG GGF GFM FMT MTS TSE SEK …
Augment word list YGGFMTSEKSQTPLVTLFKNAIIKNAHKKGQ YGG GGF GFM FMT MTS TSE SEK … AAA AAB AAC … YYY
G G F A A A 0 + 0 + -2 = -2 Non-match BLOSUM62 scores G G F G G Y 6 + 6 + 3 = 15 Match A user-specified threshold determines which three-letter words are considered matches and non-matches.
YGGFMTSEKSQTPLVTLFKNAIIKNAHKKGQ YGG GGF GFM FMT MTS TSE SEK … GGI GGL GGM GGF GGW GGY …
Store words in search tree Augmented list of query words “Does this query contain GGF?” Search tree O(1) time “Yes, at position 2.”
Search tree G G F L M W Y
Scan the database x x x x Query sequence x x x x Database sequence
Extend hit L P P Q G L L Query sequence M P P E G L L Database sequence <word> 7 2 6 BLOSUM62 scores word score = 15 <--- ---> 2 7 7 2 6 4 4 HSP SCORE = 32 hit Extend This is done by extending a hit in both directions, until the running alignment’s score has dropped more than Xbelow
BLAST 2.0The two-hit method • BLAST 1.0 • Extension step typically accounts for >90% of BLAST’ execution time • Observations: • A HSP of interest is much longer than a single word pair • Entail multiple hits on the same diagonal and within short distance of one another • Invoke an extension only when two non-overlapping hits are found within distance A on the same diagonal
Extend! < A > A • Recent[i]: the most recent hit found on the ith diagonal (always increasing) overlap
T must to be lowered • one-hits : W=3 ,T=13 • Two-hit : W=3 ,T=11 • More one-hits while the majority are dismissed • Sensitivity • For HSPs with at least 33 bits, the two-hit heuristic is more sensitive • Speed(two-hit): • Generates on average ~3.2 times as many hit, but only ~0.14 times as many hit extension(decide whether a hit need be extended) • Twice as rapid as one-hit
Gapped alignment • Original BLAST: find several distinct HSPs • All HSPs related to one alignment should be found • Gapped BLAST: tolerate a much higher chance of missing any single moderately scoring HSP • Seeking a single gapped alignment, rather than a collection of umgapped ones • For example, result should > 0.95, p: miss prob of HSP • Orignial with 2 HSP: (1-p)(1-p)>0.95 p<0.025 • Now: p2<0.05p=0.22 • T can be raised faster • Now: • Find one HSP only– seed, than use 2-hit
Gapped alignment (contd) • A gapped extension takes much longer to execute than an ungapped extension, but by performing very few of them the fraction of the total time could be kept low. • Trigger a gapped extension for any HSP exceeding score Sg • Sg should be set at ~22 bits (1:50)
Original BLAST locates only the first and the last ungapped aligment, E-value > 50 times
http://binfo.ym.edu.tw/post/internet/gap_blast.htm Gapped Local Alignments
actaactattacagactaactattacagactaactataca actaactattacggactaacttacagactaactaaaca Before Gap Insertion actaactattacagactaactattacagactaactataca |||||||||||| |||||||| | | | | actaactattacggactaacttacagactaactaaaca Percent Identity = 24/40 = 0.6 After Gap Insertion actaactattacagactaactattacagactaactataca |||||||||||| |||||||| ||||||||||||| ||| actaactattacggactaact--tacagactaactaaaca Percent Identity = 36/40 = 0.9
Gapped Local Alignments • Start from a single aligned pair of residues, called the seed.
Gapped expansion • Find out ungapped region with highest alignment score. • If the length of the ungapped region larger than Sg, then try using DP • Use its central residue pair as the seed. • Gapped extension is invoked less than once per 50 database sequences.
conserved regions • same protein family • some regions are very similar • the structure and functionality typical to this family
PSI-BLAST(Position-Specific Iterated BLAST) [1] Select a query and search it against a protein database [2] PSI-BLAST constructs a multiple sequence alignment then creates a “profile” or specialized position-specific scoring matrix (PSSM) [3] The PSSM is used as a query against the database [4] PSI-BLAST estimates statistical significance (E values) [5] Repeat steps [3] and [4] iteratively, typically 5 times. At each new search, a new profile is used as the query. PSSM PSSM From: http://bioweb.pasteur.fr/seqanal/blast/intro-uk.html
Score matrix architecture • Each matrix has length precisely equal to that of the original query sequence.
Multiple alignment construction • E-value < 0.01 from the output of BLAST output. • Any row identical to the query segment with which it aligns is purged. • Only one copy is retained of any rows that are above 98% identical to one another.
Multiple alignment construction • Pairwise alignment columns that involve gap characters inserted into the query are simply ignored. • So M has exactly the same length as the query.
Multiple alignment construction • The matrix scores for a given alignment column should depand not only upon the residues appearing there. • The set R of sequences it includes to be exactly those that contribute a residue to column C. • The columns of MC to be just those columns of M in which all the sequences of R are represented.
Sequence weights • A large set of closely related sequences carries little more information than a single member, but its size may allow it outvote a small number of more divergent sequences. • One way is to assign weights. • Gap characters are treated as a 21st distinct char.
Sequence weights • In constructing matrix scores, not only a column’s observed residue frequencies are important. • Estimate the relative number NC of independent observations constituted by the alignment MC. • NC: the mean number of different residue types.
a large number of independent sequences, the estimate of Qi should converge simply to the observed frequency of residue i in that column. • Pseudocount frequencies • Estimate Qi by:
Gapped BLAST: 1. 3X faster than original BLAST, finds more 2. >100X faster than S-W, misses only 8, same scores PSI-BLAST: 1. faster than original BLAST, 40X faster than S-W, much more sensitive 2. multiple iterations is even better, better for non-redundant database of NCBI 3. slower than gapped BLAST: time for construction of PSSM
PSI-BLAST Examples(1) 1. 二者已被證明結構相似,但用HIT當作query, a BLAST search of SWISS-PROT reveals hits with E<0.01 only to other HIT proteins. 2. A PSI-BLAST search, using PSSM generated by yields the E-value of 2X10-4for uridylyltransferase.
Seven recent additions to the protein databases as members of BRCT superfamily
Possible future improvement Gap costs • Allows a gap to involve residues in both sequences rather than just one • A gap in which k residues are inserted or deleted and j pairs of residues are left unaligned receives the score –(a+bk+cj)
Possible future improvementRealignment • 不將所有超過threshold的pairwise alignment組合成單一multiple alignment,而是只選出the most significant建構initial multiple alignment and PSSM,然後再以此rescore and realign database sequences that received lower scores • 優點 • Improve weaker pairwise alignments • False positive can be downgraded by an improved matrix • False negative can have their scores increased
Conclusion • Gapped version of BLAST is faster than original one, and able to produce gapped alignments. • PSI-BLAST greatly increase sensitivity to weak but biologically relevant sequence relationships. • PSI-BLAST retains the ability to report accurate statistics, per iteration runs in times not much greater than gapped BLAST, and can be used both iteratively and fully automatically.
NCBI • Books • Pudmed • Blast (1)Nucleotide -- Quickly search for highly similar sequences -- Nucleotide-nucleotide BLAST (2)Protein -- Protein-protein BLAST (3)Translated -- Translated query vs. Protein database (4)Special -- Align two sequences