Download
1 / 18
180 likes | 332 Views
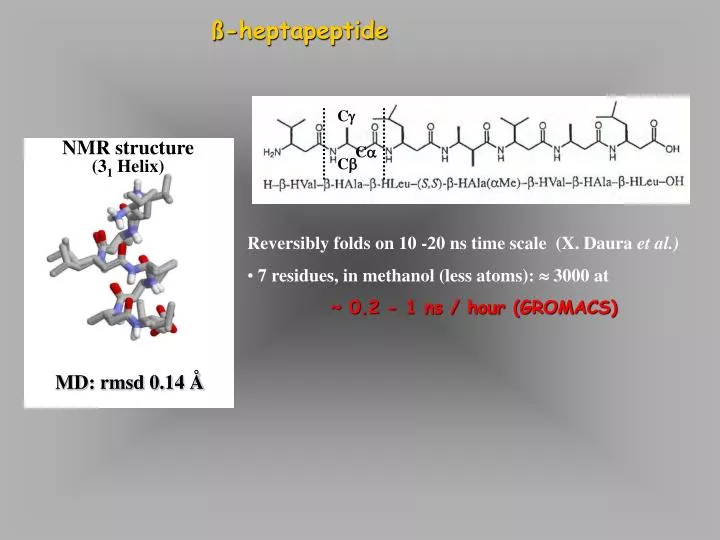
NMR structure (3 1 Helix). MD: rmsd 0.14 Å. ß-heptapeptide. C . C . C . Reversibly folds on 10 -20 ns time scale (X. Daura et al.) 7 residues, in methanol (less atoms): 3000 at ~ 0.2 - 1 ns / hour (GROMACS). ß-heptapeptide. - amino-acids (additional backbone carbon)

E N D
NMR structure (31 Helix) MD: rmsd 0.14 Å ß-heptapeptide C C C • Reversibly folds on 10 -20 ns time scale (X. Daura et al.) • 7 residues, in methanol (less atoms): 3000 at • ~ 0.2 - 1 ns / hour (GROMACS)
ß-heptapeptide • -amino-acids (additional backbone carbon) • Stable 2nd structure. • Non-degradable peptide mimetics • (e.g. highly selective somatastatin analogue) -Heptapeptide (M) 31-helix in MeOH at 298 K (left-handed) D. Seebach, B. Jaun + coworkers organic chem ETH-Zurich Daura, X., Bernhard, J., Seebach, D., van Gunsteren, W. F. and Mark, A. E. (1998) J. Mol. Biol. 280, 925-932.
ß-heptapeptide -Heptapeptide, 340 K unfold fold unfold fold unfold
ß-heptapeptide Starting structure -Heptapeptide, 360 K
ß-heptapeptide: Conventional MD • Protocol for MD: • ß-heptatpetide 7 residues in 986 methanol molecules (~3000 atoms) • GROMOS96 force field (van Gunsteren and co-workers) • GROMACS software (www.gromacs.org) • twin-range cutoff (1.0/1.4nm) for vdW and Elec. • Reaction Field for long-range Elec. • NP1atmT and NV300KT ensembles. • Temp= 275.3, 286.4, 297.8, 322.1, 348.6, 399.6K • Berendsen Thermostat, τT=0.1ps and τP=1.0ps • run on 8 processors for 1ns/hour.
Folded = rmsd from NMR Model < 0.7 Å ß-heptapeptide: Conventional MD Time (ns)
ß-heptapeptide: Conventional MD Folded = rmsd from NMR Model < 0.7 Å
ß-heptapeptide: Replica Exchange Replica Exchange Methodology
ß-heptatpetide 7 residues in 986 methanol molecules (3000 atoms) • 50 ns at 400 K to unfold it and generate random structures • 20 structures selected as initial structures for the REMD • (more than 1ns spaced and rmsd from NMR > 0.3 nm) • 200 ps of equilibration of each replica at its initial temperature • T= 275.3, 280.8, 286.8, 292.0, 297.8, 303.7, 309.7, 315.8, 322.1, 328.1, 335.0, • 341.6, 348.4, 355.4, 362.3, 369.5, 376.8, 384.2, 391.8, 399.6 • T controled by a Berendsen bath, τT=0.1ps. • V = Cste = V (NPT at 300K) • exchange trials every 0.1, 0.5, 2.0 and 5.0 ps ß-heptapeptide: Replica Exchange Protocol for the REMD:
ß-heptapeptide: Replica Exchange RMSD from the NMR model (Ca res. 2-6) Exchange trial every 2.0 ps
Ratio of Folded Peptide at each temperature Criterion: folded = rmsd 0.7Å ß-heptapeptide: Replica Exchange RMSD from the NMR model (Ca res. 2-6) 0.5ps 2.0ps 5.0ps Time of Equilibration Increasing with Time Interval Between Exchange Trial
ß-heptapeptide: Replica Exchange Folded Peptide ratios as a function of the interval between exchanges
ß-heptapeptide: Replica Exchange Temperature Exploration 0.5ps 2.0ps 5.0ps The replicas explore temperatures at different rates.
ß-heptapeptide: Replica Exchange Folded Peptide ratios as function of the interval between exchanges Simulations converged
ß-heptapeptide: Replica Exchange Folded Peptide ratios: REMD vs. Standard MD
ß-heptapeptide: Replica Exchange Folding free energy: REMD vs. Brute Force MD
322 348 T (K) 275 286 298 399 1 2 3 4 1 2 3 4 1 2 3 4 1 2 3 4 1 2 3 4 1 2 3 4 Cluster # 50 15 9 3 40 13 9 2 55 9 4 4 28 14 7 3 23 8 5 3 NPT 72 9 3 2 50159 3 40139 2 5594 4 2814 7 3 2385 3 7293 2 48 17 8 3 70 6 3 2 31 12 7 3 26 10 8 5 8 7 4 4 NVT 60 10 3 2 48178 3 7063 2 31127 3 26108 5 874 4 60103 2 65 12 7 2 61 11 8 2 43 10 7 3 29 8 7 4 13 5 5 5 REMD 69 13 7 2 65127 2 61118 2 43107 3 2987 4 1355 5 69137 2 REMD simulations reproduce conformational ensembles ß-heptapeptide: Replica Exchange Conformational exploration: REMD vs. Brute Force MD Cluster Analysis: use of the 4 more populated clusters (% of the total ensemble)
ß-heptapeptide: Replica Exchange Efficiency: REMD vs. Brute Force MD REMD simulation: 1) accurately reproduces - thermodynamics (folding free energy) - conformational (clusters) MD data for folding 2) faster than MD simulation time REMD: 20*15ns = 300ns BFMD: 2*200+400+3*800= 3200ns one Temp REMD: 20*15ns = 300ns BFMD: 1*800ns = 800ns real time REMD: 15ns / 1 CPU = 7.5 days BFMD: 800ns / 8 CPUs = 34 days

