Download
1 / 27
390 likes | 971 Views
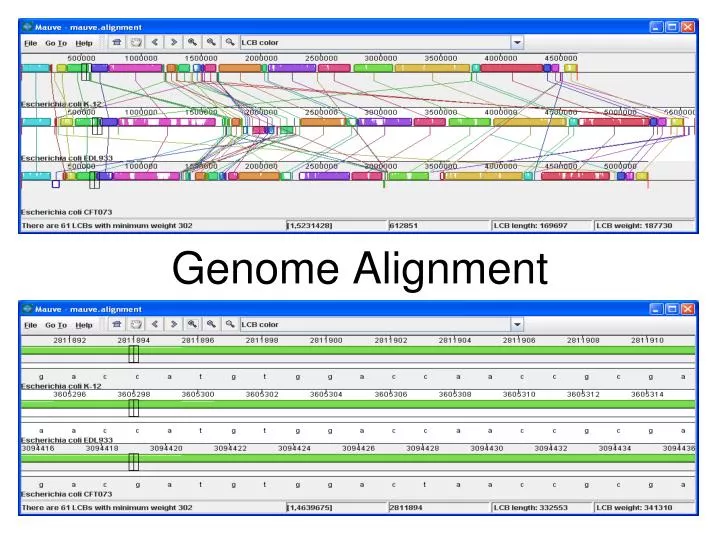
Genome Alignment. Alignment Methods. Needleman-Wunsch (global) and Smith-Waterman (local) use dynamic programming Guaranteed to find an optimal alignment given a particular scoring function Too computationally intensive for genome alignment, especially multiple genomes. Genome Alignment.

E N D
Alignment Methods • Needleman-Wunsch (global) and Smith-Waterman (local) use dynamic programming • Guaranteed to find an optimal alignment given a particular scoring function • Too computationally intensive for genome alignment, especially multiple genomes
Genome Alignment • Depending on level of similarity, genome alignments may need to contend with rearrangements and large-scale duplications and deletions • Draft or partial genomes can both benefit from and confound alignment • Need to visualize results in summary form
Genome Alignment • Pair-wise • Align two genomes • Example: MUMmer • Multiple or complex samples and a reference genome • All of one genome plus whatever parts match from the other genome(s) • Example: PIPs • Multiple alignment • All of all the genomes • Example: Mauve
Some aligners http://mummer.sourceforge.net/ MUMmer http://www.ebi.ac.uk/~bjp/pecan/Pecan http://asap.ahabs.wisc.edu/mauve/index.php Mauve
MUMmer (Maximal Unique Match) http://mummer.sourceforge.net/ • Fast pair-wise comparison of draft or complete genomes using nucleotide or 6-frame translated sequences • MUMmer 3.0 can find all 20-basepair or longer exact matches between a pair of 5-megabase genomes in 13.7 seconds, using 78 MB of memory, on a 2.4 GHz Linux desktop computer
Suffix Tree Delcher et al. Fast algorithms for large-scale genome alignment and comparison. Nucleic Acids Res. 2002 Jun 1;30(11):2478-83.
MUMMER plot Genome 2 Genome 1
5 Campylobacter PROmer analysis • Fouts et al. Major structural differences and novel potential virulence mechanisms from the genomes of multiple campylobacter species. PLoS Biol. 2005 Jan;3(1):e15. • One genome is used as the x-axis for all four pair-wise comparisons • X-shape characteristic of collinearity interrupted by inversions around the origin or terminus of replication • Loss of collinearity in more distant comparisons
Human Gut metagenomePercent Identity Plot (PIP) of random shotgun reads to a complete Bifidobacterium genome and a good quality draft Methanobrevibacter genomeGill et al. Metagenomic analysis of the human distal gut microbiome. Science. 2006 Jun 2; 312(5778): 1355-9.
Mauve Multiple Genome Aligner • Able to identify and align collinear regions of multiple genomes even in the presence of rearrangements • Find and extend seed matches • Group into locally collinear blocks • Align intervening regions • Darling et al. Genome Res. 2004 Jul;14(7):1394-403.
Progressive Mauve alignment of 12 E. coli genome Aaron Darling 2006 Ph.D. thesis,http://gel.ahabs.wisc.edu/~darling/darling_thesis.pdf
Figure 1. The difference between positional homology alignment and glocal alignment. Three example linear genomes are broken into genes labeled A,B,C,D, and R. R is a multi-copy (repetitive) gene, with different copies labeled using numeric subscripts. Each copy of R is assumed to be identical in sequence, so that orthology/paralogy is unknowable from nucleotide substitution (as is often the case with mobile DNA repeat elements). Genes shifted downward in a given genome are inverted (reverse complement) relative to the reference genome. The positional homology alignment would ideally create two local alignment blocks where each block has exactly one alignment row for each genome. Only positionally-conserved copies of the repetitive gene family R become aligned to each other. The glocal alignment would ideally create four local alignment blocks wherein all copies of the repetitive gene family become aligned to each other.
Progressive Genome Alignment similar to CLUSTAL (next week) with integrated synteny mapping and positional homology and anchored alignment
actual \ predicted negative positive Negative TN FP Positive FN TP Performance Metrics • Accuracy – Proportion correct • TN+TP/total • TPR (Recall) – Proportion of predicted positives that are correct • TP/FP+TP • Sensitivity – Proportion of positives correctly predicted • TP/FN+TP • Specificity – Proportion of negatives correctly predicted • TN/TN+FP
For nucleotide pairs, a TP is a pair aligned in both the calculated and correct alignments. A FP is a nucleotide pair in the calculated alignment that is absent from the correct alignment. Likewise, a FN is a pair in the correct alignment not present in the calculated alignment. We do not quantify True Negative (TN) alignments as the number of TN possibilities is extremely large, growing with the product of sequence lengths. Sensitivity Positive Predictive Value (PPV)
ENCODE project • Goal = to identify all functional elements in the human genome • Margulies et al. 2007 reports results of the pilot project to analyze 1% of the genome using genome alignment to detect which regions of the sequence are evolutionarily constrained.
4 aligners • MAVID • MLAGAN • TBA • PECAN • 23 mammalian species • 30 Mb; 44 regions
Alignment Coverage • For example, vs. armadillo: • MAVID 27.4% • MLAGAN 42.4% • TBA 41.2% • PECAN 40.1% • 17.4% covered by all 4 aligners • Of which 66.1% are aligned identically
Performance Metrics • Sensitivity – • coverage of protein coding regions and ancestral repeats • Specificity – • primate specific repeats (Alu) and periodicity of substitutions in protein coding regions