Download
1 / 1
10 likes | 160 Views
Dynamics differences of SAM-I riboswitch aptamer between SAM bound

E N D
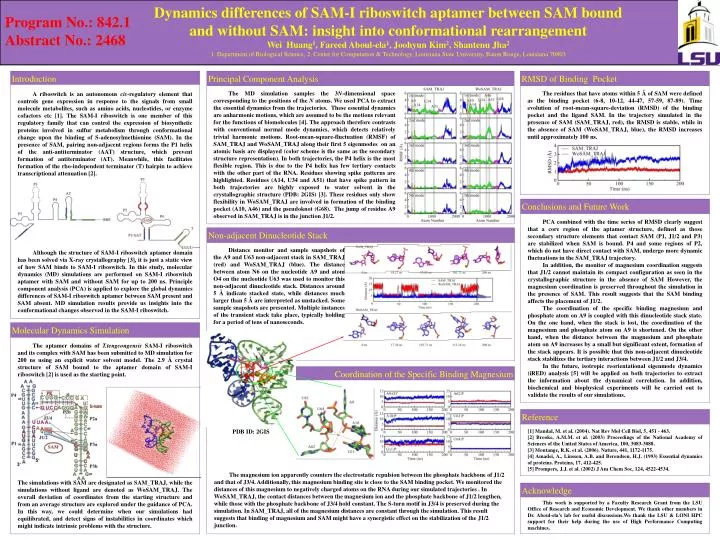
Dynamics differences of SAM-I riboswitch aptamer between SAM bound and without SAM: insight into conformational rearrangementWei Huang1, Fareed Aboul-ela1, Joohyun Kim2, Shantenu Jha21. Department of Biological Science, 2. Center for Computation & Technology, Louisiana State University, Baton Rouge, Louisiana 70803 Program No.: 842.1 Abstract No.: 2468 Reference Non-adjacent Dinucleotide Stack Acknowledge Coordination of the Specific Binding Magnesium Conclusions and Future Work Molecular Dynamics Simulation Introduction A riboswitch is an autonomous cis-regulatory element that controls gene expression in response to the signals from small molecule metabolites, such as amino acids, nucleotides, or enzyme cofactors etc [1]. The SAM-I riboswitch is one member of this regulatory family that can control the expression of biosynthetic proteins involved in sulfur metabolism through conformational change upon the binding of S-adenosylmethionine (SAM). In the presence of SAM, pairing non-adjacent regions forms the P1 helix of the anti-antiterminator (AAT) structure, which prevent formation of antiterminator (AT). Meanwhile, this facilitates formation of the rho-independent terminator (T) hairpin to achieve transcriptional attenuation [2]. Although the structure of SAM-I riboswitch aptamer domain has been solved via X-ray crystallography [3], it is just a static view of how SAM binds to SAM-I riboswitch. In this study, molecular dynamics (MD) simulations are performed on SAM-I riboswitch aptamer with SAM and without SAM for up to 200 ns. Principle component analysis (PCA) is applied to explore the global dynamics differences of SAM-I riboswitch aptamer between SAM present and SAM absent. MD simulation results provide us insights into the conformational changes observed in the SAM-I riboswitch. RMSD of Binding Pocket Principal Component Analysis The aptamer domains of T.tengcongensis SAM-I riboswitch and its complex with SAM has been submitted to MD simulation for 200 ns using an explicit water solvent model. The 2.9 Å crystal structure of SAM bound to the aptamer domain of SAM-I riboswitch [2] is used as the starting point. The simulations with SAM are designated as SAM_TRAJ, while the simulations without ligand are denoted as WoSAM_TRAJ. The overall deviation of coordinates from the starting structure and from an average structure are explored under the guidance of PCA. In this way, we could determine when our simulations had equilibrated, and detect signs of instabilities in coordinates which might indicate intrinsic problems with the structure. The residues that have atoms within 5 Å of SAM were defined as the binding pocket (6-8, 10-12, 44-47, 57-59, 87-89). Time evolution of root-mean-square-deviation (RMSD) of the binding pocket and the ligand SAM. In the trajectory simulated in the presence of SAM (SAM_TRAJ, red), the RMSD is stable, while in the absence of SAM (WoSAM_TRAJ, blue), the RMSD increases until approximately 100 ns. The magnesium ion apparently counters the electrostatic repulsion between the phosphate backbone of J1/2 and that of J3/4. Additionally, this magnesium binding site is close to the SAM binding pocket. We monitored the distances of this magnesium to negatively charged atoms on the RNA during our simulated trajectories . In WoSAM_TRAJ, the contact distances between the magnesium ion and the phosphate backbone of J1/2 lengthen, while those with the phosphate backbone of J3/4 hold constant. The S-turn motif in J3/4 is preserved during the simulation. In SAM_TRAJ, all of the magnesium distances are constant through the simulation. This result suggests that binding of magnesium and SAM might have a synergistic effect on the stabilization of the J1/2 junction. . This work is supported by a Faculty Research Grant from the LSU Office of Research and Economic Development. We thank other members in Dr. Aboul-ela’s lab for useful discussions.We thank the LSU & LONI HPC support for their help during the use of High Performance Computing machines. PCA combined with the time series of RMSD clearly suggest that a core region of the aptamer structure, defined as those secondary structure elements that contact SAM (P1, J1/2 and P3) are stabilized when SAM is bound. P4 and some regions of P2, which do not have direct contact with SAM, undergo more dynamic fluctuations in the SAM_TRAJ trajectory. In addition, the monitor of magnesium coordination suggests that J1/2 cannot maintain its compact configuration as seen in the crystallographic structure in the absence of SAM However, the magnesium coordination is preserved throughout the simulation in the presence of SAM. This result suggests that the SAM binding affects the placement of J1/2. The coordination of the specific binding magnesium and phosphate atom on A9 is coupled with this dinucleotide stack state. On the one hand, when the stack is lost, the coordination of the magnesium and phosphate atom on A9 is shortened. On the other hand, when the distance between the magnesium and phosphate atom on A9 increases by a small but significant extent, formation of the stack appears. It is possible that this non-adjacent dinucleotide stack stabilizes the tertiary interactions between J1/2 and J3/4. In the future, isotropic reorientational eigenmode dynamics (iRED) analysis [5] will be applied on both trajectories to extract the information about the dynamical correlation. In addition, biochemical and biophysical experiments will be carried out to validate the results of our simulations. [1] Mandal, M. et al. (2004). Nat Rev Mol Cell Biol, 5, 451 - 463. [2] Brooke, A.M.M. et al. (2003) Proceedings of the National Academy of Sciences of the United States of America, 100, 3083-3088. [3] Montange, R.K. et al. (2006). Nature, 441, 1172-1175. [4] Amadei, A., Linssen, A.B. and Berendsen, H.J. (1993) Essential dynamics of proteins. Proteins, 17, 412-425. [5] Prompers, J.J. et al. (2002) J Am Chem Soc, 124, 4522-4534. The MD simulation samples the 3N-dimensional space corresponding to the positions of the N atoms. We used PCA to extract the essential dynamics from the trajectories. These essential dynamics are anharmonic motions, which are assumed to be the motions relevant for the functions of biomolecules [4]. The approach therefore contrasts with conventional normal mode dynamics, which detects relatively trivial harmonic motions. Root-mean-square-fluctuation (RMSF) of SAM_TRAJ and WoSAM_TRAJ along their first 5 eigenmodes on an atomic basis are displayed (color scheme is the same as the secondary structure representation). In both trajectories, the P4 helix is the most flexible region. This is due to the P4 helix has few tertiary contacts with the other part of the RNA. Residues showing spike patterns are highlighted. Residues (A14, U34 and A51) that have spike pattern in both trajectories are highly exposed to water solvent in the crystallographic structure (PDB: 2GIS) [3]. These residues only show flexibility in WoSAM_TRAJ are involved in formation of the binding pocket (A10, A46) and the pseudoknot (G68). The jump of residue A9 observed in SAM_TRAJ is in the junction J1/2. Distance monitor and sample snapshots of the A9 and U63 non-adjacent stack in SAM_TRAJ (red) and WoSAM_TRAJ (blue). The distance between atom N6 on the nucleotide A9 and atom O4 on the nucleotide U63 was used to monitor this non-adjacent dinucleotide stack. Distances around 5 Å indicate stacked state, while distances much larger than 5 Å are interpreted as unstacked. Some sample snapshots are presented. Multiple instances of the transient stack take place, typically holding for a period of tens of nanoseconds. PDB ID: 2GIS